A Pangolin Genome Hub for the Research Community
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

71St Annual Meeting Society of Vertebrate Paleontology Paris Las Vegas Las Vegas, Nevada, USA November 2 – 5, 2011 SESSION CONCURRENT SESSION CONCURRENT
ISSN 1937-2809 online Journal of Supplement to the November 2011 Vertebrate Paleontology Vertebrate Society of Vertebrate Paleontology Society of Vertebrate 71st Annual Meeting Paleontology Society of Vertebrate Las Vegas Paris Nevada, USA Las Vegas, November 2 – 5, 2011 Program and Abstracts Society of Vertebrate Paleontology 71st Annual Meeting Program and Abstracts COMMITTEE MEETING ROOM POSTER SESSION/ CONCURRENT CONCURRENT SESSION EXHIBITS SESSION COMMITTEE MEETING ROOMS AUCTION EVENT REGISTRATION, CONCURRENT MERCHANDISE SESSION LOUNGE, EDUCATION & OUTREACH SPEAKER READY COMMITTEE MEETING POSTER SESSION ROOM ROOM SOCIETY OF VERTEBRATE PALEONTOLOGY ABSTRACTS OF PAPERS SEVENTY-FIRST ANNUAL MEETING PARIS LAS VEGAS HOTEL LAS VEGAS, NV, USA NOVEMBER 2–5, 2011 HOST COMMITTEE Stephen Rowland, Co-Chair; Aubrey Bonde, Co-Chair; Joshua Bonde; David Elliott; Lee Hall; Jerry Harris; Andrew Milner; Eric Roberts EXECUTIVE COMMITTEE Philip Currie, President; Blaire Van Valkenburgh, Past President; Catherine Forster, Vice President; Christopher Bell, Secretary; Ted Vlamis, Treasurer; Julia Clarke, Member at Large; Kristina Curry Rogers, Member at Large; Lars Werdelin, Member at Large SYMPOSIUM CONVENORS Roger B.J. Benson, Richard J. Butler, Nadia B. Fröbisch, Hans C.E. Larsson, Mark A. Loewen, Philip D. Mannion, Jim I. Mead, Eric M. Roberts, Scott D. Sampson, Eric D. Scott, Kathleen Springer PROGRAM COMMITTEE Jonathan Bloch, Co-Chair; Anjali Goswami, Co-Chair; Jason Anderson; Paul Barrett; Brian Beatty; Kerin Claeson; Kristina Curry Rogers; Ted Daeschler; David Evans; David Fox; Nadia B. Fröbisch; Christian Kammerer; Johannes Müller; Emily Rayfield; William Sanders; Bruce Shockey; Mary Silcox; Michelle Stocker; Rebecca Terry November 2011—PROGRAM AND ABSTRACTS 1 Members and Friends of the Society of Vertebrate Paleontology, The Host Committee cordially welcomes you to the 71st Annual Meeting of the Society of Vertebrate Paleontology in Las Vegas. -

Resolving the Relationships of Paleocene Placental Mammals
Biol. Rev. (2015), pp. 000–000. 1 doi: 10.1111/brv.12242 Resolving the relationships of Paleocene placental mammals Thomas J. D. Halliday1,2,∗, Paul Upchurch1 and Anjali Goswami1,2 1Department of Earth Sciences, University College London, Gower Street, London WC1E 6BT, U.K. 2Department of Genetics, Evolution and Environment, University College London, Gower Street, London WC1E 6BT, U.K. ABSTRACT The ‘Age of Mammals’ began in the Paleocene epoch, the 10 million year interval immediately following the Cretaceous–Palaeogene mass extinction. The apparently rapid shift in mammalian ecomorphs from small, largely insectivorous forms to many small-to-large-bodied, diverse taxa has driven a hypothesis that the end-Cretaceous heralded an adaptive radiation in placental mammal evolution. However, the affinities of most Paleocene mammals have remained unresolved, despite significant advances in understanding the relationships of the extant orders, hindering efforts to reconstruct robustly the origin and early evolution of placental mammals. Here we present the largest cladistic analysis of Paleocene placentals to date, from a data matrix including 177 taxa (130 of which are Palaeogene) and 680 morphological characters. We improve the resolution of the relationships of several enigmatic Paleocene clades, including families of ‘condylarths’. Protungulatum is resolved as a stem eutherian, meaning that no crown-placental mammal unambiguously pre-dates the Cretaceous–Palaeogene boundary. Our results support an Atlantogenata–Boreoeutheria split at the root of crown Placentalia, the presence of phenacodontids as closest relatives of Perissodactyla, the validity of Euungulata, and the placement of Arctocyonidae close to Carnivora. Periptychidae and Pantodonta are resolved as sister taxa, Leptictida and Cimolestidae are found to be stem eutherians, and Hyopsodontidae is highly polyphyletic. -
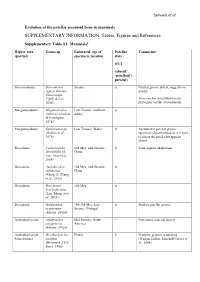
SUPPLEMENTARY INFORMATION: Tables, Figures and References
Samuels et al. Evolution of the patellar sesamoid bone in mammals SUPPLEMENTARY INFORMATION: Tables, Figures and References Supplementary Table S1: Mammals$ Higher taxa Genus sp. Estimated. age of Patellar Comments# (partial) specimen, location state 0/1/2 (absent/ ‘patelloid’/ present) Sinoconodonta Sinoconodon Jurassic 0 Patellar groove absent, suggests no rigneyi (Kielan- patella Jaworowska, Cifelli & Luo, Sinoconodon is included on our 2004) phylogeny within tritylodontids. Morganucodonta Megazostrodon Late Triassic, southern 0 rudnerae (Jenkins Africa & Parrington, 1976) Morganucodonta Eozostrodon sp. Late Triassic, Wales 0 Asymmetric patellar groove, (Jenkins et al., specimens disarticulated so it is hard 1976) to assess the patella but appears absent Docodonta Castorocauda 164 Mya, mid-Jurassic, 0 Semi-aquatic adaptations lutrasimilis (Ji, China Luo, Yuan et al., 2006) Docodonta Agilodocodon 164 Mya, mid-Jurassic, 0 scansorius China (Meng, Ji, Zhang et al., 2015) Docodonta Docofossor 160 Mya 0 brachydactylus (Luo, Meng, Ji et al., 2015) Docodonta Haldanodon 150-155 Mya, Late 0 Shallow patellar groove exspectatus Jurassic, Portugal (Martin, 2005b) Australosphenida Asfaltomylos Mid-Jurassic, South ? Postcranial material absent patagonicus America (Martin, 2005a) Australosphenida Ornithorhynchus Extant 2 Platypus, genome sequenced Monotremata anatinus (Warren, Hillier, Marshall Graves et (Herzmark, 1938; al., 2008) Rowe, 1988) Samuels et al. Australosphenida Tachyglossus + Extant 2 Echidnas Monotremata Zaglossus spp. (Herzmark, 1938; Rowe, 1988) Mammaliaformes Fruitafossor 150 Mya, Late Jurassic, 0 Phylogenetic status uncertain indet. windscheffeli (Luo Colorado & Wible, 2005) Mammaliaformes Volaticotherium Late Jurassic/Early ? Hindlimb material incomplete indet. antiquus (Meng, Cretaceous Hu, Wang et al., 2006) Eutriconodonta Jeholodens 120-125 Mya, Early 0 Poorly developed patellar groove jenkinsi (Ji, Luo Cretaceous, China & Ji, 1999) Eutriconodonta Gobiconodon spp. -

Mammalia) of São José De Itaboraí Basin (Upper Paleocene, Itaboraian), Rio De Janeiro, Brazil
The Xenarthra (Mammalia) of São José de Itaboraí Basin (upper Paleocene, Itaboraian), Rio de Janeiro, Brazil Lílian Paglarelli BERGQVIST Departamento de Geologia/IGEO/CCMN/UFRJ, Cidade Universitária, Rio de Janeiro/RJ, 21949-940 (Brazil) [email protected] Érika Aparecida Leite ABRANTES Departamento de Geologia/IGEO/CCMN/UFRJ, Cidade Universitária, Rio de Janeiro/RJ, 21949-940 (Brazil) Leonardo dos Santos AVILLA Departamento de Geologia/IGEO/CCMN/UFRJ, Cidade Universitária, Rio de Janeiro/RJ, 21949-940 (Brazil) and Setor de Herpetologia, Museu Nacional/UFRJ, Quinta da Boa Vista, Rio de Janeiro/RJ, 20940-040 (Brazil) Bergqvist L. P., Abrantes É. A. L. & Avilla L. d. S. 2004. — The Xenarthra (Mammalia) of São José de Itaboraí Basin (upper Paleocene, Itaboraian), Rio de Janeiro, Brazil. Geodiversitas 26 (2) : 323-337. ABSTRACT Here we present new information on the oldest Xenarthra remains. We conducted a comparative morphological analysis of the osteoderms and post- cranial bones from the Itaboraian (upper Paleocene) of Brazil. Several osteo- derms and isolated humeri, astragali, and an ulna, belonging to at least two species, compose the assemblage. The bone osteoderms were assigned to KEY WORDS Mammalia, Riostegotherium yanei Oliveira & Bergqvist, 1998, for which a revised diagno- Xenarthra, sis is presented. The appendicular bones share features with some “edentate” Cingulata, Riostegotherium, taxa. Many of these characters may be ambiguous, however, and comparison Astegotheriini, with early Tertiary Palaeanodonta reveals several detailed, derived resem- Palaeanodonta, blances in limb anatomy. This suggests that in appendicular morphology, one armadillo, osteoderm, of the Itaboraí Xenarthra may be the sister-taxon or part of the ancestral stock appendicular skeleton. -

Download Full Article 2.6MB .Pdf File
Memoirs of Museum Victoria 74: 151–171 (2016) Published 2016 ISSN 1447-2546 (Print) 1447-2554 (On-line) http://museumvictoria.com.au/about/books-and-journals/journals/memoirs-of-museum-victoria/ Going underground: postcranial morphology of the early Miocene marsupial mole Naraboryctes philcreaseri and the evolution of fossoriality in notoryctemorphians ROBIN M. D. BECK1,*, NATALIE M. WARBURTON2, MICHAEL ARCHER3, SUZANNE J. HAND4, AND KENNETH P. APLIN5 1 School of Environment & Life Sciences, Peel Building, University of Salford, Salford M5 4WT, UK and School of Biological, Earth and Environmental Sciences, University of New South Wales, Sydney, NSW 2052, Australia ([email protected]) 2 School of Veterinary and Life Sciences, Murdoch University, 90 South Street, Murdoch, WA 6150, Australia ([email protected]) 3 School of Biological, Earth and Environmental Sciences, University of New South Wales, Sydney, NSW 2052, Australia ([email protected]) 4 School of Biological, Earth and Environmental Sciences, University of New South Wales, Sydney, NSW 2052, Australia ([email protected]) 5 National Museum of Natural History, Division of Mammals, Smithsonian Institution, Washington, DC 20013-7012, USA ([email protected]) * To whom correspondence should be addressed. E-mail: [email protected] Abstract Beck, R.M.D., Warburton, N.M., Archer, M., Hand, S.J. and Aplin, K.P. 2016. Going underground: postcranial morphology of the early Miocene marsupial mole Naraboryctes philcreaseri and the evolution of fossoriality in notoryctemorphians. Memoirs of Museum Victoria 74: 151–171. We present the first detailed descriptions of postcranial elements of the fossil marsupial mole Naraboryctes philcreaseri (Marsupialia: Notoryctemorphia), from early Miocene freshwater limestone deposits in the Riversleigh World Heritage Area, northwestern Queensland. -

Brief Report 547
Ankle structure in Eocene pholidotan mammal Eomanis krebsi and its taxonomic implications INÉS HOROVITZ, GERHARD STORCH, and THOMAS MARTIN Georges Cuvier (1798) established the classical concept of teristics they found it shared with Eo. waldi. On the other hand, E. Edentata which included sloths, anteaters, armadillos, aard− joresi was considered to be a myrmecophagid anteater, order varks, and pangolins. With the growing body of comparative Xenarthra, becoming the only representative of this order outside morphological data becoming available during the nine− of the Americas, and the oldest anteater in the fossil record. Alter− teenth century, it was evident that Cuvier's “Edentata” was native hypotheses for the evolutionary affinities of E. joresi have an artificial group (e.g., Huxley 1872). In his classical text− been proposed by other authors, and some of the more detailed book, Weber (1904) excluded aardvarks and pangolins from analyses have suggested a sister group relationship with Pilosa the Edentata and put them in separate orders, Tubulidentata (a subgroup of Xenarthra consisting of anteaters and sloths) and Pholidota. Later on, fossil taxa were repeatedly added to (Gaudin and Branham 1998), or a position outside of Xenarthra, and removed from Edentata, such as various xenarthran such as a close relationship with the extinct Palaeanodonta (Rose groups, taeniodonts, palaeanodonts, and gondwanatheres, 1999), or indeterminate (Szalay and Schrenk 1998). but the South American Xenarthra always was considered as Szalay and Schrenk (1998) contended that similarities be− their core group. Even the living order Pholidota has been tween Eomanis krebsi and Eurotamandua joresi were larger in cited again as ?Edentata incertae sedis many years after number and importance than those between Eo. -

The Morphology of Xenarthrous Vertebrae (Mammalia: Xenarthra)
FIELDIANA GEOLOGY LIBRAE Geology NEW SERIES, NO. 41 The Morphology of Xenarthrous Vertebrae (Mammalia: Xenarthra) Timothy J. Gaudin en 2z September 30, 1999 Publication 1505 PUBLISHED BY FIELD MUSEUM OF NATURAL HISTORY Information for Contributors to Fieldiana General: Fieldiana is primarily a journal for Field Museum staff members and research associates, although manuscripts from nonaffiliated authors may be considered as space permits. The Journal carries a page charge of $65.00 per printed page or fraction thereof. Payment of at least 50% of page charges qualifies a paper for expedited processing, which reduces the publication time. Contributions from staff, research associates, and invited authors will be considered for publication regardless of ability to pay page charges, however, the full charge is mandatory for nonaffiliated authors of unsolicited manuscripts. Three complete copies of the text (including title page and abstract) and of the illustrations should be submitted (one original copy plus two review copies which may be machine copies). No manuscripts will be considered for publication or submitted to reviewers before all materials are complete and in the hands of the Scientific Editor. Manuscripts should be submitted to Scientific Editor, Fieldiana, Field Museum of Natural History, Chicago, Illinois 60605-2496, U.S.A. Text: Manuscripts must be typewritten double-spaced on standard-weight, 8Vi- by 11 -inch paper with wide margins on all four sides. If typed on an IBM-compatible computer using MS-DOS, also submit text on S^-inch diskette (WordPerfect 4.1, 4.2, or 5.0, MultiMate, Displaywrite 2, 3 & 4, Wang PC, Samna, Microsoft Word, Volks- writer, or WordStar programs or ASCII). -

The University of Michigan
CONTRIBUTIONS FROM THE MUSEUM OF PALEONTOLOGY THE UNIVERSITY OF MICHIGAN VOL. 28. No. 10, PP. 221-24s December 15. 1992 SKELETON OF ALOCODONTULUM ATOPUM, AN EARLY EOCENE EPOICOTHERIID (MAMMALIA, PALAEANODONTA) FROM THE BIGIIORN BASIN, WYOh4ING KENNETH D. ROSE, ROBERT J. EMRY, AND PHILIP D. GINGERICH MUSEUM OF PALEONTOLOGY THE UNIVERSITY OF MICHIGAN ANN ARBOR CONTRIBUTIONS FROM THE MUSEUM OF PALEONTOLOGY Philip D. Gingerich, Director This series of contributions from the Museum of Paleontology is a medium for publication of papers based chiefly on collections in the Museum. When the number of pages issued is sufficient to make a volume, a title page and a table of contents will be sent to libraries on the mailing list, and to individuals on request. A list of the separate issues may also be obtained by request. Correspondenceshould be directed to the Museum of Paleontology, The University of Michigan, Ann Arbor, Michigan 48109-1079. VOLS. 2-28. Parts of volumes may be obtained if available. Price lists are available upon inquiry. SKELETON OF ALOCODONTULUM ATOPUM, AN EARLY EOCENE EPOICOTHERIID (MAMMALIA, PALAEANODONTA) FROM THE BIGHORN BASIN, WYOMING BY KENNETH D. ROSE', ROBERT J. EMRY~,AND PHILIP D. GINGERICH Abstract.- A substantially complete skeleton of the early Eocene palaeanodont Alocodontulum atopum from the Bighorn Basin, Wyoming, is described. It is the oldest and most complete known skeleton referable to the family Epoicotheriidae. Alocodontulum was dentally more generalized but post- cranially more specialized than the contemporary metacheiromyid Palaeano- don. It displays numerous modifications for fossorial habits, which are particularly prevalent in the forelimb skeleton. Certain characters, especially in the manus, foreshadow specializations carried to extreme in subterranean Oligocene epoicotheriids. -

The Evolution of Armadillos, Anteaters and Sloths Depicted by Nuclear and Mitochondrial Phylogenies: Implications for the Status of the Enigmatic Fossil Eurotamandua
The evolution of armadillos, anteaters and sloths depicted by nuclear and mitochondrial phylogenies: implications for the status of the enigmatic fossil Eurotamandua. Frédéric Delsuc, François Catzeflis, Michael Stanhope, Emmanuel Douzery To cite this version: Frédéric Delsuc, François Catzeflis, Michael Stanhope, Emmanuel Douzery. The evolution of armadil- los, anteaters and sloths depicted by nuclear and mitochondrial phylogenies: implications for the status of the enigmatic fossil Eurotamandua.. Proceedings of the Royal Society B: Biological Sciences, Royal Society, The, 2001, 268 (1476), pp.1605-15. 10.1098/rspb.2001.1702. halsde-00192975 HAL Id: halsde-00192975 https://hal.archives-ouvertes.fr/halsde-00192975 Submitted on 30 Nov 2007 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Final version accepted for publication in The Proceedings of the Royal Society of London B [20.04.2001] Main text = 4753 words ______________________________________________________________________ The evolution of armadillos, anteaters, and sloths depicted by nuclear and mitochondrial phylogenies: implications for the status of the enigmatic fossil Eurotamandua . Frédéric DELSUC 1* , François M. CATZEFLIS 1, Michael J. STANHOPE 2 and Emmanuel J. P. DOUZERY 1 1 Laboratoire de Paléontologie, Paléobiologie et Phylogénie, Institut des Sciences de l’Evolution, Université Montpellier II, Montpellier, France 2 Biology and Biochemistry, Queens University, 97 Lisburn Road, Belfast BT9 7BL, UK. -
Download a High Resolution
DENVER MUSEUM OF NATURE & SCIENCE ANNALS NUMBER 1, SEPTEMBER 1, 2009 Land Mammal Faunas of North America Rise and Fall During the Early Eocene Climatic Optimum Michael O. Woodburne, Gregg F. Gunnell, and Richard K. Stucky WWW.DMNS.ORG/MAIN/EN/GENERAL/SCIENCE/PUBLICATIONS The Denver Museum of Nature & Science Annals is an Frank Krell, PhD, Editor-in-Chief open-access, peer-reviewed scientific journal publishing original papers in the fields of anthropology, geology, EDITORIAL BOARD: paleontology, botany, zoology, space and planetary Kenneth Carpenter, PhD (subject editor, Paleontology and sciences, and health sciences. Papers are either authored Geology) by DMNS staff, associates, or volunteers, deal with DMNS Bridget Coughlin, PhD (subject editor, Health Sciences) specimens or holdings, or have a regional focus on the John Demboski, PhD (subject editor, Vertebrate Zoology) Rocky Mountains/Great Plains ecoregions. David Grinspoon, PhD (subject editor, Space Sciences) Frank Krell, PhD (subject editor, Invertebrate Zoology) The journal is available online at www.dmns.org/main/en/ Steve Nash, PhD (subject editor, Anthropology and General/Science/Publications free of charge. Paper copies Archaeology) are exchanged via the DMNS Library exchange program ([email protected]) or are available for purchase EDITORIAL AND PRODUCTION: from our print-on-demand publisher Lulu (www.lulu.com). Betsy R. Armstrong: project manager, production editor DMNS owns the copyright of the works published in the Ann W. Douden: design and production Annals, which are published under the Creative Commons Faith Marcovecchio: proofreader Attribution Non-Commercial license. For commercial use of published material contact Kris Haglund, Alfred M. Bailey Library & Archives ([email protected]). -

New Postcranial Bones of the Extinct Mammalian Family Nyctitheriidae (Paleogene, UK): Primitive Euarchontans with Scansorial Locomotion
Palaeontologia Electronica palaeo-electronica.org New postcranial bones of the extinct mammalian family Nyctitheriidae (Paleogene, UK): Primitive euarchontans with scansorial locomotion Jerry J. Hooker ABSTRACT New postcranial bones from the Late Eocene and earliest Oligocene of the Hamp- shire Basin, UK, are identified as belonging to the extinct family Nyctitheriidae. Previ- ously, astragali and calcanea were the only elements apart from teeth and jaws to be unequivocally recognized. Now, humeri, radii, a metacarpal, femora, distal tibiae, naviculars, cuboids, an ectocuneiform, metatarsals and phalanges, together with addi- tional astragali and calcanea, have been collected. These are shown to belong to a diversity of nyctithere taxa previously named from dental remains. Functional analysis shows that nyctitheres had mobile shoulder and hip joints, could pronate and supinate the radius, partially invert the foot at the astragalocalcaneal and upper ankle joints using powerful flexor muscles, all indicative of a scansorial lifestyle and allowing head- first descent on vertical surfaces. Climbing appears to have been dominated by flexion of the forearms and feet. Cladistic analysis employing a range of primitive eutherian mammals shows that nyctitheres are stem euarchontans, rather than lipotyphlans, with which they had previously been classified based on dental characters. Earlier ideas of relationships with the extinct Adapisoriculidae, recently considered stem eutherians, are not upheld. Jerry J. Hooker. Department of Earth Sciences, Natural History Museum, Cromwell Road, London SW7 5BD, UK. [email protected] Keywords: Eutheria; Lipotyphla; placental; Scandentia; shrews; Soricidae INTRODUCTION basis of some straight slender limb bone shafts, apparently associated with teeth (on which this Nyctitherium Marsh, 1872, the type genus of genus was based), returned to Marsh’s original the eutherian family Nyctitheriidae Simpson, 1928a idea that Nyctitherium was a chiropteran. -

Nonmarine Bivalves from the Lower Permian (Wolfcampian) of the Chama Basin, New Mexico Spencer G
New Mexico Geological Society Downloaded from: http://nmgs.nmt.edu/publications/guidebooks/56 Nonmarine bivalves from the Lower Permian (Wolfcampian) of the Chama Basin, New Mexico Spencer G. Lucas and Larry F. Rinehart, 2005, pp. 283-287 in: Geology of the Chama Basin, Lucas, Spencer G.; Zeigler, Kate E.; Lueth, Virgil W.; Owen, Donald E.; [eds.], New Mexico Geological Society 56th Annual Fall Field Conference Guidebook, 456 p. This is one of many related papers that were included in the 2005 NMGS Fall Field Conference Guidebook. Annual NMGS Fall Field Conference Guidebooks Every fall since 1950, the New Mexico Geological Society (NMGS) has held an annual Fall Field Conference that explores some region of New Mexico (or surrounding states). Always well attended, these conferences provide a guidebook to participants. Besides detailed road logs, the guidebooks contain many well written, edited, and peer-reviewed geoscience papers. These books have set the national standard for geologic guidebooks and are an essential geologic reference for anyone working in or around New Mexico. Free Downloads NMGS has decided to make peer-reviewed papers from our Fall Field Conference guidebooks available for free download. Non-members will have access to guidebook papers two years after publication. Members have access to all papers. This is in keeping with our mission of promoting interest, research, and cooperation regarding geology in New Mexico. However, guidebook sales represent a significant proportion of our operating budget. Therefore, only research papers are available for download. Road logs, mini-papers, maps, stratigraphic charts, and other selected content are available only in the printed guidebooks.