The 2007 WHO Classification of Tumours of the Central Nervous System
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Risk Factors for Gliomas and Meningiomas in Males in Los Angeles County1
[CANCER RESEARCH 49, 6137-6143. November 1, 1989] Risk Factors for Gliomas and Meningiomas in Males in Los Angeles County1 Susan Preston-Martin,2 Wendy Mack, and Brian E. Henderson Department of Preventive Medicine, University of Southern California School of Medicine, Los Angeles, California 90033 ABSTRACT views with proxy respondents, we were unable to include a large proportion of otherwise eligible cases because they were deceased or Detailed job histories and information about other suspected risk were too ill or impaired to participate in an interview. The Los Angeles factors were obtained during interviews with 272 men aged 25-69 with a County Cancer Surveillance Program identified the cases (26). All primary brain tumor first diagnosed during 1980-1984 and with 272 diagnoses had been microscopically confirmed. individually matched neighbor controls. Separate analyses were con A total of 478 patients were identified. The hospital and attending ducted for the 202 glioma pairs and the 70 meningioma pairs. Meningi- physician granted us permission to contact 396 (83%) patients. We oma, but not glioma, was related to having a serious head injury 20 or were unable to locate 22 patients, 38 chose not to participate, and 60 more years before diagnosis (odds ratio (OR) = 2.3; 95% confidence were aphasie or too ill to complete the interview. We interviewed 277 interval (CI) = 1.1-5.4), and a clear dose-response effect was observed patients (74% of the 374 patients contacted about the study or 58% of relating meningioma risk to number of serious head injuries (/' for trend the initial 478 patients). -

Original Article Outcome of Supratentorial Intraaxial Extra Ventricular Primary Pediatric Brain Tumors
Original Article Outcome of supratentorial intraaxial extra ventricular primary pediatric brain tumors: A prospective study Mohana Rao Patibandla, Suchanda Bhattacharjee1, Megha S. Uppin2, Aniruddh Kumar Purohit1 Department of Neurosurgery, Krishna Institute of Medical Sciences Secunderabad, Departments of 1Neurosurgery and 2Pathology, Nizam’s Institute of Medical Sciences, Hyderabad, Andhra Pradesh, India Address for correspondence: Dr. Mohana Rao Patibandla, Department of Neurosurgery, University of Colorado Denver, 13123, E 16th Ave, Aurora, CO 80045, USA. Email: [email protected] ABSTRACT Introduction: Tumors of the central nervous system (CNS) are the second most frequent malignancy of childhood and the most common solid tumor in this age group. CNS tumors represent approximately 17% of all malignancies in the pediatric age range, including adolescents. Glial neoplasms in children account for up to 60% of supratentorial intraaxial tumors. Their histological distribution and prognostic features differ from that of adults. Aims and Objectives: To study clinical and pathological characteristics, and to analyze the outcome using the Engel’s classification for seizures, Karnofsky’s score during the available follow‑up period of minimum 1 year following the surgical and adjuvant therapy of supratentorial intraaxial extraventricular primary pediatric (SIEPP) brain tumors in children equal or less than 18 years. Materials and Methods: The study design is a prospective study done in NIMS from October 2008 to January 2012. All the patients less than 18 years of age operated for SIEPP brain tumors proven histopathologically were included in the study. All the patients with recurrent or residual primary tumors or secondaries were excluded from the study. Post operative CT or magnetic resonance imaging (MRI) is done following surgery. -

Charts Chart 1: Benign and Borderline Intracranial and CNS Tumors Chart
Charts Chart 1: Benign and Borderline Intracranial and CNS Tumors Chart Glial Tumor Neuronal and Neuronal‐ Ependymomas glial Neoplasms Subependymoma Subependymal Giant (9383/1) Cell Astrocytoma(9384/1) Myyppxopapillar y Desmoplastic Infantile Ependymoma Astrocytoma (9412/1) (9394/1) Chart 1: Benign and Borderline Intracranial and CNS Tumors Chart Glial Tumor Neuronal and Neuronal‐ Ependymomas glial Neoplasms Subependymoma Subependymal Giant (9383/1) Cell Astrocytoma(9384/1) Myyppxopapillar y Desmoplastic Infantile Ependymoma Astrocytoma (9412/1) (9394/1) Use this chart to code histology. The tree is arranged Chart Instructions: Neuroepithelial in descending order. Each branch is a histology group, starting at the top (9503) with the least specific terms and descending into more specific terms. Ependymal Embryonal Pineal Choro id plexus Neuronal and mixed Neuroblastic Glial Oligodendroglial tumors tumors tumors tumors neuronal-glial tumors tumors tumors tumors Pineoblastoma Ependymoma, Choroid plexus Olfactory neuroblastoma Oligodendroglioma NOS (9391) (9362) carcinoma Ganglioglioma, anaplastic (9522) NOS (9450) Oligodendroglioma (9390) (9505 Olfactory neurocytoma Ganglioglioma, malignant (()9521) anaplastic (()9451) Anasplastic ependymoma (9505) Olfactory neuroepithlioma Oligodendroblastoma (9392) (9523) (9460) Papillary ependymoma (9393) Glioma, NOS (9380) Supratentorial primitive Atypical EdEpendymo bltblastoma MdllMedulloep ithliithelioma Medulloblastoma neuroectodermal tumor tetratoid/rhabdoid (9392) (9501) (9470) (PNET) (9473) tumor -

Central Nervous System Tumors General ~1% of Tumors in Adults, but ~25% of Malignancies in Children (Only 2Nd to Leukemia)
Last updated: 3/4/2021 Prepared by Kurt Schaberg Central Nervous System Tumors General ~1% of tumors in adults, but ~25% of malignancies in children (only 2nd to leukemia). Significant increase in incidence in primary brain tumors in elderly. Metastases to the brain far outnumber primary CNS tumors→ multiple cerebral tumors. One can develop a very good DDX by just location, age, and imaging. Differential Diagnosis by clinical information: Location Pediatric/Young Adult Older Adult Cerebral/ Ganglioglioma, DNET, PXA, Glioblastoma Multiforme (GBM) Supratentorial Ependymoma, AT/RT Infiltrating Astrocytoma (grades II-III), CNS Embryonal Neoplasms Oligodendroglioma, Metastases, Lymphoma, Infection Cerebellar/ PA, Medulloblastoma, Ependymoma, Metastases, Hemangioblastoma, Infratentorial/ Choroid plexus papilloma, AT/RT Choroid plexus papilloma, Subependymoma Fourth ventricle Brainstem PA, DMG Astrocytoma, Glioblastoma, DMG, Metastases Spinal cord Ependymoma, PA, DMG, MPE, Drop Ependymoma, Astrocytoma, DMG, MPE (filum), (intramedullary) metastases Paraganglioma (filum), Spinal cord Meningioma, Schwannoma, Schwannoma, Meningioma, (extramedullary) Metastases, Melanocytoma/melanoma Melanocytoma/melanoma, MPNST Spinal cord Bone tumor, Meningioma, Abscess, Herniated disk, Lymphoma, Abscess, (extradural) Vascular malformation, Metastases, Extra-axial/Dural/ Leukemia/lymphoma, Ewing Sarcoma, Meningioma, SFT, Metastases, Lymphoma, Leptomeningeal Rhabdomyosarcoma, Disseminated medulloblastoma, DLGNT, Sellar/infundibular Pituitary adenoma, Pituitary adenoma, -

Choroid Plexus Tumors: a Review
UC San Diego UC San Diego Previously Published Works Title Perinatal (fetal and neonatal) choroid plexus tumors: a review. Permalink https://escholarship.org/uc/item/0sm7q5q7 Journal Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery, 35(6) ISSN 0256-7040 Authors Crawford, John R Isaacs, Hart Publication Date 2019-06-01 DOI 10.1007/s00381-019-04135-x Peer reviewed eScholarship.org Powered by the California Digital Library University of California Child's Nervous System (2019) 35:937–944 https://doi.org/10.1007/s00381-019-04135-x REVIEW ARTICLE Perinatal (fetal and neonatal) choroid plexus tumors: a review John R. Crawford1,2,3 & Hart Isaacs Jr3,4 Received: 13 September 2018 /Accepted: 20 March 2019 /Published online: 5 April 2019 # Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract Introduction The object of this review is to describe the choroid plexus tumors (CPTs) occurring in the fetus and neonate with regard to clinical presentation, location, pathology, treatment, and outcome. Materials and methods Case histories and clinical outcomes were reviewed from 93 cases of fetal and neonatal tumors obtained from the literature and our own institutional experience from 1980 to 2016. Results Choroid plexus papilloma (CPP) is the most common tumor followed by choroid plexus carcinoma (CPC) and atypical choroid plexus papilloma (ACPP). Hydrocephalus and macrocephaly are the presenting features for all three tumors. The lateral ventricles are the main site of tumor origin followed by the third and fourth ventricles, respectively. CPTs of the fetus are detected most often near the end of the third trimester of pregnancy by fetal ultrasound. -

Risk-Adapted Therapy for Young Children with Embryonal Brain Tumors, High-Grade Glioma, Choroid Plexus Carcinoma Or Ependymoma (Sjyc07)
SJCRH SJYC07 CTG# - NCT00602667 Initial version, dated: 7/25/2007, Resubmitted to CPSRMC 9/24/2007 and 10/6/2007 (IRB Approved: 11/09/2007) Activation Date: 11/27/2007 Amendment 1.0 dated January 23, 2008, submitted to CPSRMC: January 23, 2008, IRB Approval: March 10, 2008 Amendment 2.0 dated April 16, 2008, submitted to CPSRMC: April 16, 2008, (IRB Approval: May 13, 2008) Revision 2.1 dated April 29, 2009 (IRB Approved: April 30, 2009 ) Amendment 3.0 dated June 22, 2009, submitted to CPSRMC: June 22, 2009 (IRB Approved: July 14, 2009) Activated: August 11, 2009 Amendment 4.0 dated March 01, 2010 (IRB Approved: April 20, 2010) Activated: May 3, 2010 Amendment 5.0 dated July 19, 2010 (IRB Approved: Sept 17, 2010) Activated: September 24, 2010 Amendment 6.0 dated August 27, 2012 (IRB approved: September 24, 2012) Activated: October 18, 2012 Amendment 7.0 dated February 22, 2013 (IRB approved: March 13, 2013) Activated: April 4, 2013 Amendment 8.0 dated March 20, 2014. Resubmitted to IRB May 20, 2014 (IRB approved: May 22, 2014) Activated: May 30, 2014 Amendment 9.0 dated August 26, 2014. (IRB approved: October 14, 2014) Activated: November 4, 2014 Un-numbered revision dated March 22, 2018. (IRB approved: March 27, 2018) Un-numbered revision dated October 22, 2018 (IRB approved: 10-24-2018) RISK-ADAPTED THERAPY FOR YOUNG CHILDREN WITH EMBRYONAL BRAIN TUMORS, HIGH-GRADE GLIOMA, CHOROID PLEXUS CARCINOMA OR EPENDYMOMA (SJYC07) Principal Investigator Amar Gajjar, M.D. Division of Neuro-Oncology Department of Oncology Section Coordinators David Ellison, M.D., Ph.D. -
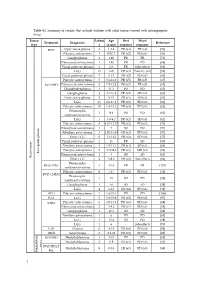
Table S1. Summary of Studies That Include Children with Solid Tumors Treated with Antiangiogenic Drugs
Table S1. Summary of studies that include children with solid tumors treated with antiangiogenic drugs. Tumor Patient Age Best Worst Treatment Diagnostic Reference type s (years) response response BVZ Optic nerve glioma 4 1.2-4 PR (x4) PR (x4) [56] Pilocytic astrocytoma 3 0.92-7 PR (x2) PD (x1) [56] Ganglioglioma 1 1.92 PR PR [56] Pilomyxoid astrocytoma 1 1.92 PR PD [68] Visual pathway glioma 1 2.6 PR Side effects [66] LGG 15 1-20 CR (x3) Toxicity (x1) [58] Visual pathway glioma 3 6-13 PR (x2) PD (x1) [60] Pilocytic astrocytoma 5 3.1-11.2 PR (x5) PR (x5) [65] BVZ+IRO Pilomyxoid astrocytoma 2 7.9-12.2 PR (x2) PR (x2) [65] Oligodendroglioma 1 11.1 PD PD [65] Ganglioglioma 3 4.1-16.8 PR (x2) SD (x1) [65] Optic nerve glioma 2 9-15 PR (x1) SD (x1) [63] LGG 35 0.6-17.6 PR (x2) PD (x8) [61] Pilocytic astrocytoma 10 1.8-15.3 PR (x4) PD (x1) [62] Pleomorphic 1 9.4 PD PD [62] xanthoastrocytoma LGG 5 3.9-9.2 PR (x2) SD (x3) [62] Pilocytic astrocytoma 4 4.67-12.17 PR (x2) PD (x3) [57] Pilomyxoid astrocytoma 1 3 SD PD [57] Fibrillary astrocytoma 3 1.92-11.08 CR (x1) PD (x3) [57] Other LGG 6 1-13.42 PR (x2) PD (x6) [57] Visual pathway glioma 1 11 PR SD [60] Fibrillary astrocytoma 3 1.5-11.1 CR (x1) SD (x1) [64] Pilocytic astrocytoma 2 3.75-9.8 PR (x1) MR (x1) [64] Low-grade glioma Pilomyxoid astrocytoma 1 3 SD SD [64] Brain tumor Brain tumor Other LGG 4 3-9.6 PR (x2) Side effects [64] Pleomorphic BVZ+TMZ 1 13.4 PR PR [153] xanthoastrocytoma Pilocytic astrocytoma 9 5-17 PR (x3) PD (x3) [59] BVZ+CHEM Pleomorphic 1 18 SD PD [59] xanthoastrocytoma Ganglioglioma -

Losses of Chromosomal Arms 1P and 19Q in the Diagnosis of Oligodendroglioma
Losses of Chromosomal Arms 1p and 19q in the Diagnosis of Oligodendroglioma. A Study of Paraffin- Embedded Sections Peter C. Burger, M.D., A. Yuriko Minn, M.S., Justin S. Smith, M.D., Ph.D., Thomas J. Borell, B.S., Anne E. Jedlicka, M.S., Brenda K. Huntley, B.S., Patricia T. Goldthwaite, M.S., Robert B. Jenkins, M.D., Ph. D., Burt G. Feuerstein, M.D., Ph.D. The Departments of Pathology (PCB, PTG) and Anesthesiology and Critical Care Medicine (AEJ), Johns Hopkins University School of Medicine, Baltimore, Maryland; Department of Pathology and Laboratory Medicine, Mayo Clinic (JSS, TJB, BKH, RBJ), Rochester, Minnesota; and Departments of Laboratory Medicine and Neurosurgery and Brain Tumor Research Center, University of California at San Francisco School of Medicine (BGF, AYM), San Francisco, California It is the impression of some pathologists that many Comparative genomic hybridization (CGH), fluores- gliomas presently designated as oligodendrogliomas cence in situ hybridization (FISH), polymerase chain would have been classified in the past as astrocyto- reaction–based microsatellite analysis, and p53 se- mas with little thought about the possibility of an quencing were performed in paraffin-embedded ma- oligodendroglial component. It also appears that the terial from 18 oligodendrogliomas and histologically relative incidence of the diagnoses of oligodendrogli- similar astrocytomas. The study was undertaken be- oma and astrocytomas varies widely from institution cause of evidence that concurrent loss of both the 1p to institution, suggesting that diagnostic criteria dif- and 19q chromosome arms is a specific marker for fer. In some laboratories, even minor or focal nuclear oligodendrogliomas. Of the six lesions with a review roundness or perinuclear haloes are considered to be diagnosis of oligodendroglioma, all had the predicted evidence of oligodendroglial differentiation, whereas loss of 1p and 19q seen by CGH, FISH, and polymerase pathologists elsewhere require the more classical fea- chain reaction. -

Molecular Insights Into Malignant Progression of Atypical Choroid Plexus Papilloma
Downloaded from molecularcasestudies.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press COLD SPRING HARBOR Molecular Case Studies | RESEARCH REPORT Molecular insights into malignant progression of atypical choroid plexus papilloma Maxim Yankelevich,1,2,9 Jonathan L. Finlay,3 Hamza Gorsi,2 William Kupsky,4 Daniel R. Boue,5 Carl J. Koschmann,6,7 Chandan Kumar-Sinha,7,8 and Rajen Mody6,7 1Pediatric Hematology/Oncology Program, Rutgers Cancer Institute of New Jersey, New Brunswick, New Jersey 08901, USA; 2Division of Pediatric Hematology/Oncology, Children’s Hospital of Michigan and Wayne State University School of Medicine, Detroit, Michigan 48201, USA; 3Division of Hematology, Oncology, and BMT, Nationwide Children’s Hospital and The Ohio State University, College of Medicine, Columbus, Ohio 43205, USA; 4Department of Pathology, Wayne State University School of Medicine, Detroit, Michigan 48201, USA; 5Department of Pathology, Nationwide Children’s Hospital and The Ohio State University, College of Medicine, Columbus, Ohio 43205, USA; 6Department of Pediatrics, Michigan Medicine, 7Rogel Cancer Center, 8Michigan Center for Translational Pathology, Michigan Medicine, University of Michigan, Ann Arbor, Michigan 48109, USA Abstract Choroid plexus tumors are rare pediatric neoplasms ranging from low-grade pap- illomas to overtly malignant carcinomas. They are commonly associated with Li–Fraumeni syndrome and germline TP53 mutations. Choroid plexus carcinomas associated with Li–Fraumeni syndrome are less responsive to chemotherapy, and there is a need to avoid radiation therapy leading to poorer outcomes and survival. Malignant progression from choroid plexus papillomas to carcinomas is exceedingly rare with only a handful of cases re- ported, and the molecular mechanisms of this progression remain elusive. -

JMSCR Vol||05||Issue||07||Page 25362-25366||July 2017
JMSCR Vol||05||Issue||07||Page 25362-25366||July 2017 www.jmscr.igmpublication.org Impact Factor 5.84 Index Copernicus Value: 83.27 ISSN (e)-2347-176x ISSN (p) 2455-0450 DOI: https://dx.doi.org/10.18535/jmscr/v5i7.174 Original Article Choroid Plexus Carcinoma of Third Ventricle – A Case Report Authors Dr N.L.N.Moorthy1, Dr S. Padmaja2, Dr M. Vithaleswar Rao3, Dr K. Jitender Reddy4, Dr B.G.Ratnam5 1Professor Radiology, 2Senior Resident Radiology, 3Assistant Professor Radiology 4Senior Consultant Radiology, 5Senior Consultant Neurosurgeon Department of Radiology, Apollo Institute of Medical Science and Research, Hyderabad 500096 Corresponding Author Dr S. Padmaja Department of Radiology, Apollo Institute of Medical Science & Research, Jubilee Hills, Hyderabad 500096 Email: [email protected], mobile 9652965345 ABSTRACT Choroid plexus carcinomas are extremely rare and rapidly growing intra ventricular tumors which are more frequently seen in young children. Choroid plexus tumors commonly involve lateral ventricles and fourth ventricle and are sometimes seen in third ventricle and extra ventricular location. We report a case of choroid plexus carcinoma arising from third ventricle extending into fourth ventricle in a 13 year old girl and discuss the imaging findings. Keywords: Choroid plexus carcinoma- third ventricle- intra ventricular tumors. INTRODUCTION tumors2. The most common site is lateral ventricle Choroid plexus is a mass of vascular structure (50 %), followed by fourth ventricle (40 %) and present in cerebral ventricular system. It is third ventricle (5 %). The extraventricular location responsible for the production and filtration of include cerebellopontine angle, suprasellar region, cerebrospinal fluid and acts as blood –CSF pineal gland and cerebellum. -

Brain and Spine Tumors
Brain and Spine Tumors Andrew J. Fabiano, MD FAANS Associate Professor of Neurosurgery Roswell Park Cancer Institute SUNY at Buffalo School of Medicine Brain Tumors Brain Tumor Basics Types of Tumors Cases Brain Tumors Skull is a fixed space Symptoms develop due to compression of normal brain Brain Tumors Brain Tumors Inflammation/Edema occurs in the surrounding normal brain Brain Tumors Tumors cause edema and irritation of normal brain Breakdown of BBB Corticosteroids for edema Anti-epileptics to prevent seizures Corticosteroids Dexamethasone traditionally used Reduces vasogenic edema GI prophylaxis Steroids Multiple side effects: Diabetes Myopathy Infection LE edema Weight gain Wound issues Anti-Epileptic Drugs Used for cortical lesions Not required for cerebellar lesions Dilantin – requires monitoring Keppra Tumor Types Gliomas Meningiomas Metastatic Tumors Pituitary Tumors Gliomas Arise from native cells within the brain Gliomas WHO I – Pilocytic Astrocytoma WHO II – Fibrillary Astrocytoma WHO III – Anaplastic Astrocytoma WHO IV – Glioblastoma Multiforme Gliomas – WHO I Gliomas – WHO II & III WHO IV - GBM Glioblastoma Multiforme Most common primary brain tumor in adults Incidence 3 per 100,000 Average survival from diagnosis ~ 13 months Young age, High Karnofsky score associated with increased survival Glioblastoma Start steroids and anti-epileptics Glioblastoma Gliomas - Treatment Surgery Biopsy External Beam XRT Chemotherapy (Temodar) Glioblastoma Survival Related to Extent of Resection Glioblastoma Typical IMRT course is Monday-Friday -

Conversion of a Gemistocytic Astrocytoma to a Fibrillary Astrocytoma After Temozolomide and Radiation Therapy
Open Access Case Report DOI: 10.7759/cureus.219 Conversion of a Gemistocytic Astrocytoma to a Fibrillary Astrocytoma after Temozolomide and Radiation Therapy Brandon C. Gabel, Mary Goolsby, Lawrence Hansen, Bob Carter, Clark Chen 1. Corresponding author: Brandon C. Gabel, [email protected] Abstract In most instances, gemistocytic astrocytoma progresses to anaplastic astrocytoma at the time of recurrence. Here, we report an unusual case where a gemistocytic astrocytoma converted to a more benign fibrillary histology after temozolomide/radiation therapy. We present the case of a 26-year-old woman with a history of fibrillary astrocytoma that was followed over a span of eight years and underwent three craniotomies. The pathology from the first craniotomy revealed a fibrillary astrocytoma. The pathology from the second craniotomy (one year after the first resection) revealed findings consistent with a gemistocytic astrocytoma. The pathology from a third craniotomy (seven years after the second resection) revealed a fibrillary astrocytoma without a significant gemistocytic component. This is the first documented case of a gemistocytic astrocytoma converting to a more benign fibrillary pathology at the time of recurrence. We speculate that the phenomenon is the result of the inherently heterogeneous astrocytic subpopulations that were differentially sensitive to chemo/radiation. Categories: Neurology, Neurosurgery, Oncology Keywords: gemistocytic, fibrillary, astrocytoma, temozolomide, radiation, glioma Introduction It is a commonly accepted truism in oncology that the grade and aggressiveness of a tumor increases at the time of recurrence. As an example of this phenomenon, recurrent gemistocytic astrocytoma nearly always exhibit less differentiated or anaplastic histology [1-2]. We present an interesting case of a gemistocytic astrocytoma that converted to a more benign fibrillary histology at the time of recurrence.