Membrane Assisted Liquid-Liquid Extraction of Cerium
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Use of Phosphate Solubilizing Bacteria to Leach Rare Earth Elements from Monazite-Bearing Ore
Minerals 2015, 5, 189-202; doi:10.3390/min5020189 OPEN ACCESS minerals ISSN 2075-163X www.mdpi.com/journal/minerals Article Use of Phosphate Solubilizing Bacteria to Leach Rare Earth Elements from Monazite-Bearing Ore Doyun Shin 1,2,*, Jiwoong Kim 1, Byung-su Kim 1,2, Jinki Jeong 1,2 and Jae-chun Lee 1,2 1 Mineral Resources Resource Division, Korea Institute of Geoscience and Mineral Resources (KIGAM), Gwahangno 124, Yuseong-gu, Daejeon 305-350, Korea; E-Mails: [email protected] (J.K.); [email protected] (B.K.); [email protected] (J.J.); [email protected] (J.L.) 2 Department of Resource Recycling Engineering, Korea University of Science and Technology, Gajeongno 217, Yuseong-gu, Daejeon 305-350, Korea * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +82-42-868-3616. Academic Editor: Anna H. Kaksonen Received: 8 January 2015 / Accepted: 27 March 2015 / Published: 2 April 2015 Abstract: In the present study, the feasibility to use phosphate solubilizing bacteria (PSB) to develop a biological leaching process of rare earth elements (REE) from monazite-bearing ore was determined. To predict the REE leaching capacity of bacteria, the phosphate solubilizing abilities of 10 species of PSB were determined by halo zone formation on Reyes minimal agar media supplemented with bromo cresol green together with a phosphate solubilization test in Reyes minimal liquid media as the screening studies. Calcium phosphate was used as a model mineral phosphate. Among the test PSB strains, Pseudomonas fluorescens, P. putida, P. rhizosphaerae, Mesorhizobium ciceri, Bacillus megaterium, and Acetobacter aceti formed halo zones, with the zone of A. -

COLUMBIUM - and RARE-EARTH ELEMENT-BEARING DEPOSITS at BOKAN MOUNTAIN, SOUTHEAST ALASKA by J
COLUMBIUM - AND RARE-EARTH ELEMENT-BEARING DEPOSITS AT BOKAN MOUNTAIN, SOUTHEAST ALASKA By J. Dean Warner and James C. Barker Alaska Field Operations Center * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * OFR - 33-89 UNITED STATES DEPARTMENT OF THE INTERIOR Manuel Lujan, Jr., Secretary BUREAU OF MINES T.S. Ary, Director TABLE OF CONTENTS Page Abstract ........................................................ 1 Introduction ....................... 2 Acknowledgments ................................................. 4 Location, access, and physiography .............................. 5 History and production ............. 5 Geologic setting .................................. 9 Trace element analyses of the peralkaline granite .............. 11 Nature of Bureau investigations ................................. 13 Sampling methods and analytical techniques .................... 13 Analytical interference, limitations, and self-shielding ...... 13 Resource estimation methods ................................... 15 Prospect evaluations .......................................... 16 Shear zones and fracture-controlled deposits .................... 18 Ross Adams mine ................. 18 Sunday Lake prospect.......................................... 21 I and L No. 1 and Wennie prospects ............................ 27 Other Occurrences ............................................. 31 Altered peralkaline granite ................................. 31 Dotson shear zone ........................................... 35 Resources .................................................. -

(12) Patent Application Publication (10) Pub. No.: US 2014/0166788 A1 Pearse Et Al
US 2014O166788A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0166788 A1 Pearse et al. (43) Pub. Date: Jun. 19, 2014 (54) METHOD AND SYSTEM FOR MAGNETIC (52) U.S. Cl. SEPARATION OF RARE EARTHIS CPC. B03C I/02 (2013.01); B02C 23/08 (2013.01); B02C23/20 (2013.01) (76) Inventors: Gary Pearse, Ottawa (CA); Jonathan USPC .......... 241/20: 209/212: 209/214; 241/24.14; Borduas, Montreal (CA); Thomas 241/21 Gervais, Montreal (CA); David Menard, Laval (CA); Djamel Seddaoui, Repentigny (CA); Bora Ung, Quebec (CA) (57) ABSTRACT (21) Appl. No.: 13/822,363 A system and a method for separating rare earth element (22) PCT Filed: Aug. 15, 2012 compounds from a slurry of mixed rare earth element com pounds, comprising flowing the slurry of mixed rare earth (86). PCT No.: PCT/CA12/50.552 element compounds through at least a first channel rigged S371 (c)(1), with at least a first magnet along a length thereof and con (2), (4) Date: May 17, 2013 nected to at least a first output channel at the position of the Publication Classification magnet, and retrieving individual rare earth element com pounds and/or groups of rare earth element compounds, sepa (51) Int. Cl. rated from the slurry as they are selectively attracted by the BO3C I/02 (2006.01) magnet and directed in the corresponding output channel B2C 23/20 (2006.01) according to their respective ratio of magnetic Susceptibility B2C 23/08 (2006.01) (AX) to specific density (Ap). Ferromagnetic particles are dia?pararnegnetic particles Submitted to a magnetic torque remain relatively unaffected by the torque As a result they roll left \\ Carried right by the slow rotating drum Patent Application Publication Jun. -

WO 2011/156817 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date Χ t it o n 1 15 December 2011 (15.12.2011) WO 2011/156817 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C02F1/58 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, (21) International Application Number: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, PCT/US20 11/040214 DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, 13 June 201 1 (13.06.201 1) KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (25) Filing Language: English NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (26) Publication Language: English SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/354,031 11 June 2010 ( 11.06.2010) (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant (for all designated States except US): MOLY- GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, CORP MINERALS LLC [US/US]; 561 Denver Tech ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, Center Pkwy, Suite 1000, Greenwood Village, CO 801 11 TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, (US). -

Some Uses of Mischmetall in Organic Synthesis
REVIEW 9 Mischmetall in organic synthesis Organic synthesis Organic synthesis New from Acros Organics Marie-Isabelle Lannou, Florence Hélion and Jean-Louis Namy Laboratoire de Catalyse Moléculaire, associé au CNRS, ICMO, Bat 420, Université de Paris-Sud, 91405, Orsay, France Some uses of mischmetall in organic synthesis ∗ Marie-Isabelle Lannou, Florence Hélion and Jean-Louis Namy Laboratoire de Catalyse Moléculaire, associé au CNRS, ICMO, Bat 420, Université Paris-Sud, 91405, Orsay, France. Abstract: Mischmetall, an alloy of the light lanthanides, has been used in a variety of organic reactions, either as a coreductant in samarium(II)-mediated reactions (Barbier and Grignard-type reactions, pinacolic coupling reactions) or as the promoter of Reformatsky-type reactions. It has been also employed as the starting material for easy syntheses of lanthanide trihalides, the reactivity of which has been explored in Imamoto and Luche-Fukuzawa reactions and in Mukaiyama aldol reactions. Keywords: mischmetall, samarium, lanthanide, catalysis, organic reactions Introduction The use of rare earth compounds in organic chemistry has grown considerably during the past twenty years. Mainly samarium, cerium, lanthanum, ytterbium, neodymium, dysprosium, lutetium, scandium and yttrium metals and derivatives have been studied. These elements clearly differ in terms of reactivity. For instance, samarium(II) compounds can be used as powerful reductants whereas cerium(IV) ones are strong oxidants, some scandium and ytterbium(III) derivatives are very efficient catalysts in a variety of reactions and chemistry of lanthanum metal shows special features. We thought however that in many reactions, a mixture of lanthanide could be used instead of individual element without significant change in results. -
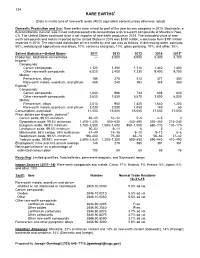
Mineral Commodity Summaries 2016
134 RARE EARTHS1 [Data in metric tons of rare-earth oxide (REO) equivalent content unless otherwise noted] Domestic Production and Use: Rare earths were mined for part of the year by one company in 2015. Bastnäsite, a fluorocarbonate mineral, was mined and processed into concentrates and rare-earth compounds at Mountain Pass, CA. The United States continued to be a net importer of rare-earth products in 2015. The estimated value of rare- earth compounds and metals imported by the United States in 2015 was $150 million, a decrease from $191 million imported in 2014. The estimated distribution of rare earths by end use was as follows, in decreasing order: catalysts, 60%; metallurgical applications and alloys, 10%; ceramics and glass, 10%; glass polishing, 10%; and other, 10%. Salient Statistics—United States: 2011 2012 2013 2014 2015e Production, bastnäsite concentrates — 3,000 5,500 5,400 4,100 Imports:2 Compounds: Cerium compounds 1,120 1,390 1,110 1,440 1,400 Other rare-earth compounds 6,020 3,400 7,330 9,400 9,700 Metals: Ferrocerium, alloys 186 276 313 371 360 Rare-earth metals, scandium, and yttrium 468 240 393 348 460 Exports:2 Compounds: Cerium compounds 1,640 996 734 608 600 Other rare-earth compounds 3,620 1,830 5,570 3,800 6,000 Metals: Ferrocerium, alloys 2,010 960 1,420 1,640 1,200 Rare-earth metals, scandium, and yttrium 3,030 2,080 1,050 140 60 Consumption, estimated 11,000 15,000 15,000 17,000 17,000 Price, dollars per kilogram, yearend:3 Cerium oxide, 99.5% minimum 40–45 10–12 5–6 4–5 2 Dysprosium oxide, 99.5% minimum 1,400–1,420 -

Bastnaesite Beneficiation by Froth Flotation and Gravity Separation
BASTNAESITE BENEFICIATION BY FROTH FLOTATION AND GRAVITY SEPARATION by Nathanael Williams A thesis submitted to the Faculty and the Board of Trustees of the Colorado School of Mines in partial fulfillment of the requirements for the degree of Master of Science (Metallurgical and Materials Engineering). Golden, Colorado Date___________ Signed:_______________________ Nathanael Williams Date___________ Signed:_______________________ Dr. Corby Anderson Thesis Advisor Date:___________ Signed:_______________________ Dr. Angus Rockett Department Head Department of Metallurgical and Materials Engineering ii ABSTRACT Rare earth elements are in high demand in the United States. Independence from the importation of rare earths is essential to alleviate dependence on China for these rare earth elements. Bastnaesite, a rare earth fluorocarbonate, is one of the most abundant sources of rare earths in the United States. It is a fluorocarbonate mineral containing primarily cerium and lanthanum. The largest rare earth mine in the United States is Mountain Pass. This research was done to find a way to combine flotation with novel collectors and gravity separation techniques to reach an enhanced grade and recovery of rare earth elements while rejecting the gangue minerals, calcite, barite and silicate minerals. The main economic driving force is the price of hydrochloric acid in downstream processes, as calcite is an acid consumer. Surface chemistry analysis was completed using adsorption density, zeta potential, and microflotation on both gravity concentrates and run of mine ore samples. Four collectors were examined. These were N,2.dihydroxybenzamide, N-hydroxycyclohexanecarboxamide, N,3. dihydroxy-2.naphthamide, and N-hydroxyoleamide. Through this analysis it was determined that, to obtain the desired results, that flotation would be the rougher stage and the gravity separation would be utilized as the cleaner stage. -

Open Application of Pourbaix Diagrams
The Pennsylvania State University The Graduate School Department of Materials Science and Engineering APPLICATION OF POURBAIX DIAGRAMS IN THE HYDROMETALLURGICAL PROCESSING OF BASTNASITE A Thesis in Materials Science and Engineering by Isehaq S. Al-Nafai © 2015 Isehaq S. Al-Nafai Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science May 2015 i The thesis of Isehaq S. Al-Nafai was reviewed and approved* by the following: Kwadwo Osseo-Asare Distinguished Professor of Materials Science and Engineering, Metallurgy and Energy and Geo-Environmental Engineering. Thesis Adviser Hojong Kim Assistant Professor of Materials Science and Engineering Ismaila Dabo Assistant Professor of Materials Science and Engineering Suzanne Mohney Professor of Materials Science and Engineering and Electrical Engineering Chair, Intercollege Graduate Degree Program in Materials Science and Engineering *Signatures are on file in the Graduate School. ii ABSTRACT The hydrometallurgical processing of bastnasite was studied by constructing Pourbaix (potential vs. pH) diagrams at room temperature using HSC Chemistry 5.0 software. Different systems were considered to understand the overall behaviors of bastnasite and its species in aqueous systems. Most of the thermodynamic data were taken from HSC database, others were collected from literature and some were estimated. The standard Gibbs free energy of formation of bastnasite, REFCO3, were estimated using four different estimation methods. Each method shows its applicability, and the obtained average values were -379.9 kcal/mol for CeFCO3, -382.3 kcal/mol for LaFCO3, -379.8 kcal/mol for NdFCO3 and -381.3 kcal/mol for PrFCO3. RE-F-CO3-H2O systems show the stability regions of bastnasite which are located in nearly neutral to alkaline media (pH ~ 6.5-11). -

Molybdenum, Ferromolybdenum, and Ammonium Molybdate, Phosphoric
MOLYBDENUM, FERROMOLYBDENUM, AND AMMONIUM MOLYBDATE A. Commodity Summary Almost all molybdenum is recovered from low-grade deposits of the mineral molybdenite, naturally occurring molybdenum disulfide (MoS2), mined either from a primary deposit, or as a byproduct of copper processing.1 In 1993, one mine extracted molybdenum ore, and nine mines recovered molybdenum as a byproduct. Two plants converted molybdenite concentrate to molybdic oxide, which was used to produced ferromolybdenum, metal powder, and other molybdenum compounds.2 Exhibit 1 presents the names and locations of molybdenum mines and processing facilities. EXHIBIT 1 SUMMARY OF MOLYBDENUM, MOLYBDIC OXIDE, AND FERROMOLYBDENUM PRODUCERSa Facility Name Location Cyprus-Climax - Henderson Empire, CO Cyprus-Climax Fort Madison, IA Cyprus-Climax Clear Water, MI Cyprus-Climax - Green Valley Tucson, AZ Cyprus-Climax Baghdad, AZ Kennecott Bingham Canyon, UT Molycorp Inc. Washington, PA Montana Resources Inc. Butte, MT Phelps Dodge Chino, NM San Manuel San Manuel, AZ San Manuel Morenci, AZ Thompson Creek Chalis, ID Thompson Creek Langeloth, PA a - Personal Communication between ICF Incorporated and John W. Blossom, U.S. Bureau of Mines, October 1994. Molybdenum metal is a refractory metal used as an alloying agent in steels, cast irons, and superalloys.3 Ferromolybdenum is an alloy of iron and molybdenum used primarily as an alternative additive in producing alloy steels, cast irons, and nonferrous alloys. The two most common grades of ferromolybdenum are low carbon- and high carbon ferromolybdenum. Ammonium molybdate is an intermediate in manufacturing both molybdenum metal and molybdic oxide, although it can also be sold as a product. Purified MoS2 concentrate also is used as a lubricant. -

Recovery of Rare Earth Elements from an Apatite Concentrate
Doctoral Thesis in Chemical Engineering Recovery of Rare Earth Elements from an Apatite Concentrate Mahmood Alemrajabi KTH Royal Institute of technology School of Engineering Sciences in Chemistry, Biotechnology and Health Stockholm, Sweden 2018 Recovery of Rare Earth Elements from an Apatite Concentrate Doctoral Thesis in Chemical Engineering 2018 Mahmood Alemrajabi TRITA-CBH-FOU-2018:49 ISBN 978-91-7873-034-6 ISSN 1654-1081 KTH Royal Institute of Technology School of Engineering Sciences in Chemistry, Biotechnology and Health Department of Chemical Engineering SE-100 44 Stockholm Paper I: Copyright 2017 Elsevier Paper II: Copyright 2018 Elsevier Tryck: Universitetsservice US-AB, Stockholm 2018 To My Love Rana IV Abstract Rare earth elements (REE) are a group of 17 elements including lanthanides, yttrium and scandium; which are found in a variety of classes of minerals worldwide. The criticality of the application, lack of high grade and economically feasible REE resources and a monopolistic supply situation has raised significant attention in recovery of these metals from low grade ores and waste materials. In this thesis, the recovery of REE from an apatite concentrate, containing 0.5 mass% of REE, within the nitrophosphate route of fertilizer production has been investigated. Most of the REE (≥ 95%) content can be recovered into a phosphate precipitate with almost 30 mass% REE. Different processes have been developed to convert the REE phosphate precipitate into a more soluble form to obtain a solution suitable for further REE purification and individual separation. It has been shown that after reprecipitation of the REE phosphate concentrate as REE sodium double sulphate and then transformation into a REE hydroxide concentrate, a solution containing 45g/L REE free of Ca, Fe and P can be obtained. -

Rare Earths1
132 RARE EARTHS1 [Data in metric tons of rare-earth oxide (REO) equivalent content unless otherwise noted] Domestic Production and Use: Rare earths were not mined domestically in 2017. Bastnaesite, a rare-earth fluorocarbonate mineral, was previously mined as a primary product at Mountain Pass, CA, which was put on care- and-maintenance status in the fourth quarter of 2015. The estimated value of rare-earth compounds and metals imported by the United States in 2017 was $150 million, a significant increase from $118 million imported in 2016. The estimated distribution of rare earths by end use was as follows: catalysts, 55%; ceramics and glass, 15%; metallurgical applications and alloys, 10%; polishing, 5%; and other, 15%. Salient Statistics—United States: 2013 2014 2015 2016 2017e Production, bastnäsite concentrates 5,500 5,400 5,900 — — Imports:2 Compounds: Cerium compounds 1,110 2,990 1,440 1,830 2,700 Other rare-earth compounds 7,330 9,260 7,720 9,650 9,300 Metals: Ferrocerium, alloys 313 371 356 269 290 Rare-earth metals, scandium, and yttrium 393 348 385 404 400 Exports:2 Compounds: Cerium compounds 734 608 440 309 220 Other rare-earth compounds 5,570 3,780 4,540 281 420 Metals: Ferrocerium, alloys 1,420 1,640 1,220 943 1,300 Rare-earth metals, scandium, and yttrium 1,050 140 60 103 140 Consumption, apparent3 5,870 12,200 9,550 10,500 11,000 Price, dollars per kilogram, yearend4 Cerium oxide, 99.5% minimum 5–6 4–5 2 2 3 Dysprosium oxide, 99.5% minimum 440–490 320–360 215–240 185–193 180–190 Europium oxide, 99.99% minimum 950–1,000 680–730 90–110 62–70 75–80 Lanthanum oxide, 99.5% minimum 6 5 2 2 3 Mischmetal, 65% cerium, 35% lanthanum 9–10 9–10 5–6 5–6 6 Neodymium oxide, 99.5% minimum 65–70 56–60 39–42 38–40 56–59 Terbium oxide, 99.99% minimum 800–850 590–640 410–470 410–425 470–480 Employment, mine and mill, annual average 380 391 351 — — Net import reliance5 as a percentage of apparent consumption 6 56 38 100 100 Recycling: Limited quantities, from batteries, permanent magnets, and fluorescent lamps. -

Rare Earth Element Phases in Bauxite Residue
minerals Article Rare Earth Element Phases in Bauxite Residue Johannes Vind 1,2,* ID , Annelies Malfliet 3, Bart Blanpain 3, Petros E. Tsakiridis 2 ID , Alan H. Tkaczyk 4 ID , Vicky Vassiliadou 1 and Dimitrios Panias 2 1 Department of Continuous Improvement and Systems Management, Aluminium of Greece Plant, Metallurgy Business Unit, Mytilineos S.A., Agios Nikolaos, 32003 Boeotia, Greece; [email protected] 2 School of Mining and Metallurgical Engineering, National Technical University of Athens, Iroon Polytechniou 9, Zografou Campus, 15780 Athens, Greece; [email protected] (P.E.T.); [email protected] (D.P.) 3 Department of Materials Engineering, KU Leuven, Kasteelpark Arenberg 44, P.O. Box 2450, B-3001 Leuven, Belgium; annelies.malfl[email protected] (A.M.); [email protected] (B.B.) 4 Institute of Physics, University of Tartu, Ostwaldi 1, 50411 Tartu, Estonia; [email protected] * Correspondence: [email protected]; Tel.: +30-210-7722184 Received: 30 January 2018; Accepted: 15 February 2018; Published: 24 February 2018 Abstract: The purpose of present work was to provide mineralogical insight into the rare earth element (REE) phases in bauxite residue to improve REE recovering technologies. Experimental work was performed by electron probe microanalysis with energy dispersive as well as wavelength dispersive spectroscopy and transmission electron microscopy. REEs are found as discrete mineral particles in bauxite residue. Their sizes range from <1 µm to about 40 µm. In bauxite residue, the most abundant REE bearing phases are light REE (LREE) ferrotitanates that form a solid solution between the phases with major compositions (REE,Ca,Na)(Ti,Fe)O3 and (Ca,Na)(Ti,Fe)O3.