Minutes of PRAC Meeting on 09 -12 March 2020
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Summary of Drug Limitations Mary C
RON DESANTIS GOVERNOR SUMMARY OF DRUG LIMITATIONS MARY C. MAYHEW SECRETARY **Medications listed in this document may or may not require a prior authorization. Please view the Preferred Drug List at: http://ahca.myflorida.com/Medicaid/Prescribed_Drug/pharm_thera/fmpdl.shtml** Summary of Drug Limitations Abilify (aripiprazole) 2mg, 5mg, 20mg, 30mg tablets Minimum age = 6; Maximum of 1 tablet per day Abilify (aripiprazole) 10mg, 15mg tablets Minimum age = 6; Maximum of 15mg per day for ages = 6 - 11; Maximum of 30mg per day for ages = 12-17 Maximum of 1 tablet per day Abilify (aripiprazole) Discmelt 10mg, 15mg tabs Minimum age = 6; Maximum of 15mg per day for ages = 6 - 11; Maximum of 30mg per day for ages = 12-17; Maximum of 2 tablets per day Abilify (aripiprazole) 1mg/ml solution Minimum age = 6; Maximum of 15ml per day for ages = 6 - 11; Maximum of 30ml per day for ages = 12-17; Maximum of 30ml per day for ages =/> 18 Abilify Maintena (aripiprazole) syringe/vial Minimum age = 18; Maximum of 1 syringe or vial every 28 days Absorica (isotretinoin) 10mg, 20mg, 25mg,30mg, 35mg, & Minimum age = 12 40mg capsules Abstral (fentanyl citrate) sublingual tablets Minimum age = 18; Maximum of 4 sublingual tablets per day Acanya (benzoyl peroxide/clindamycin)Gel, gel pump Minimum Age= 12 Accolate (zafirlukast) tablets Maximum of 3 tablets per day Aciphex (rabeprazole) 5mg, 10mg sprinkle capsules Minimum age = 1; Maximum age = 11; Maximum of 1 capsule per day Aciphex (rabeprazole) 20mg tablets Minimum age = 1; Maximum of 2 tablets per day Actemra (tocilizumab) 80mg/4ml, 200mg/10ml, Minimum age= 2 400mg/20ml Vials, & 162mg/0.9ml Syringe Actimmune (Interferon Gamma-1b) Maximum of 6ml every 28days Actiq (fentanyl citrate) Lozenges Minimum age = 18; Maximum of 4 lozenges per day Activella (estradiol/norethindrone) tablets Minimum age = 18 Updated 02/28/2019 1 RON DESANTIS GOVERNOR SUMMARY OF DRUG LIMITATIONS MARY C. -

XERMELO™ (Telotristat Ethyl)
PHARMACY COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 5/18/2017 SECTION: DRUGS LAST REVIEW DATE: 5/20/2021 LAST CRITERIA REVISION DATE: 5/20/2021 ARCHIVE DATE: XERMELO™ (telotristat ethyl) Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Pharmacy Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Pharmacy Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide BCBSAZ complete medical rationale when requesting any exceptions to these guidelines. The section identified as “Description” defines or describes a service, procedure, medical device or drug and is in no way intended as a statement of medical necessity and/or coverage. The section identified as “Criteria” defines criteria to determine whether a service, procedure, medical device or drug is considered medically necessary or experimental or investigational. State or federal mandates, e.g., FEP program, may dictate that any drug, device or biological product approved by the U.S. Food and Drug Administration (FDA) may not be considered experimental or investigational and thus the drug, device or biological product may be assessed only on the basis of medical necessity. Pharmacy Coverage Guidelines are subject to change as new information becomes available. For purposes of this Pharmacy Coverage Guideline, the terms "experimental" and "investigational" are considered to be interchangeable. BLUE CROSS®, BLUE SHIELD® and the Cross and Shield Symbols are registered service marks of the Blue Cross and Blue Shield Association, an association of independent Blue Cross and Blue Shield Plans. -

List of Medicinal Products Under Additional Monitoring
26 October 2018 EMA/245297/2013 Rev.60 Inspections & Human Medicines Pharmacovigilance List of medicinal products under additional monitoring Related Information: Additional monitoring explained: http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/document_listing/document_listing_000365.jsp Good Pharmacovigilance Practice Module on additional monitoring: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000345.jsp To note: All products added to the list in September 2018 are shown in red font. All products removed from the list are shown with a strikethrough for the period of one month after which they are excluded. Date of Product name Active Substance (s) Reason (s) on list Marketing Authorisation Holder (s) Link to Product Information Inclusion http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/ Abasaglar (previously Abasria) Insulin glargine New biological Eli Lilly Nederland B.V. 002835/human_med_001790.jsp&mid=WC0b01ac058001d124 October 2014 Acarizax (also known in some EU countries as MITIZAX) Standardised allergen extract from house dust mites New Biological ALK-Abelló A/S https://portal.dimdi.de/amispb/doc/pei/Web/2613318-spcde-20170401.pdf May 2016 http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/ Accofil Filgrastim New biological Accord Healthcare Limited 003956/human_med_001798.jsp&mid=WC0b01ac058001d124 October 2014 http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/ Adcetris Brentuximab vedotin -
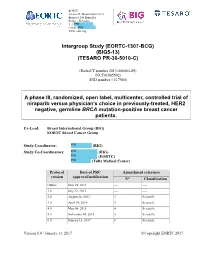
Eortc-1307-Bcg) (Big5-13) (Tesaro Pr-30-5010-C)
EORTC Avenue E. Mounierlaan 83/11 Brussel 1200 Bruxelles België – Belgique Tel: PPD e-mail: PPD www.eortc.org Intergroup Study (EORTC-1307-BCG) (BIG5-13) (TESARO PR-30-5010-C) (EudraCT number 2013-000684-85) (NCT01905592) (IND number # 117580) A phase III, randomized, open label, multicenter, controlled trial of niraparib versus physician’s choice in previously-treated, HER2 negative, germline BRCA mutation-positive breast cancer patients. Co-Lead: Breast International Group (BIG) EORTC Breast Cancer Group Study Coordinator: PPD (BIG) Study Co-Coordinators: PPD (BIG) PPD (EORTC) PPD (Tufts Medical Center) Protocol Date of PRC Amendment reference version approval/notification N° Classification Outline June 28, 2013 ---- ---- 1.0 July 22, 2013 ---- ---- 2.0 August 28, 2013 1 Scientific 3.0 April 24, 2014 3 Scientific 4.0 May 04, 2015 4 Scientific 5.0 November 04, 2015 5 Scientific 6.0 January 13, 2017 8 Scientific Version 6.0 / January 13, 2017 Copyright EORTC 2017 EORTC-1307-BCG / BIG5-13 / TESARO PR-30-5010-C Niraparib in BRCA germline mutated MBC Contact addresses Executive Committee: Defined in the related study charter Writing Committee: PPD (EORTC, Statistician) PPD (EORTC, Study Co-coordinator) PPD (Tufts Medical Center) PPD (BIG, Scientific Director) PPD (BIG, Ass. Scientific Director) PPD (EORTC, Statistician) PPD (EORTC, Clinical Research Physician) PPD (BIG, Study Coordinator) Steering Committee: Defined in the related study charter Version 6.0 2 / 102 January 13, 2017 EORTC-1307-BCG / BIG5-13 / TESARO PR-30-5010-C Niraparib -
Quantity Limits/Daily Dose Limits
Effective 07/20/21 Alphabetical by Brand Name (when applicable) PA Medical Assistance Fee-for-Service Quantity Limits & Daily Dose Limits Maximum Therapy Class Brand Name Generic Name Daily Dose Limit Antipsychotics, Atypical ABILIFY 1 MG/ML SOLUTION ARIPIPRAZOLE 25 Antipsychotics, Atypical ABILIFY 10 MG DISCMELT ARIPIPRAZOLE 2 Antipsychotics, Atypical ABILIFY 10 MG TABLET ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY 15 MG DISCMELT ARIPIPRAZOLE 2 Antipsychotics, Atypical ABILIFY 15 MG TABLET ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY 2 MG TABLET ARIPIPRAZOLE 2 Antipsychotics, Atypical ABILIFY 20 MG TABLET ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY 30 MG TABLET ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY 5 MG TABLET ARIPIPRAZOLE 1.5 Antipsychotics, Atypical ABILIFY 9.75 MG/1.3 ML INJECTION VIAL ARIPIPRAZOLE 3.9 Antipsychotics, Atypical ABILIFY MAINTENA ER 300 MG SYRINGE ARIPIPRAZOLE 0.04 Antipsychotics, Atypical ABILIFY MAINTENA ER 300 MG VIAL ARIPIPRAZOLE 0.04 Antipsychotics, Atypical ABILIFY MAINTENA ER 400 MG SYRINGE ARIPIPRAZOLE 0.04 Antipsychotics, Atypical ABILIFY MAINTENA ER 400 MG VIAL ARIPIPRAZOLE 0.04 Antipsychotics, Atypical ABILIFY MYCITE 10 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 15 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 2 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 20 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 30 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 5 MG KIT ARIPIPRAZOLE 1 Antivirals, Herpes ABREVA 10% -

Mississippi Division of Medicaid Universal Preferred Drug List
MISSISSIPPI DIVISION OF MEDICAID EFFECTIVE 01/01/2021 UNIVERSAL PREFERRED DRUG LIST Version 2021.13a Updated: 8-30-2021 (For All Medicaid, MSCAN and CHIP Beneficiaries) Conduent’s SmartPA Pharmacy Application (SmartPA) is a proprietary electronic prior authorization system used for Medicaid fee for service claims. MSCAN plans may/may not -have electronic PA functionality. However, they must adhere to Medicaid’s PA criteria. THERAPEUTIC PREFERRED AGENTS NON-PREFERRED AGENTS PA CRITERIA DRUG CLASS ACNE AGENTS ANTI-INFECTIVE clindamycin gel (generic Cleocin-T) ACZONE (dapsone) Maximum Age Limit clindamycin lotion AKNE-MYCIN (erythromycin) • 21 years – all agents except clindamycin solution azelaic acid isotretinoins AMZEEQ FOAM (minocycline) AZELEX (azelaic acid) CLEOCIN-T (clindamycin) CLINDAMYCIN PAC (clindamycin) CLINDAGEL (clindamycin) clindamycin foam clindamycin gel daily (generic Clindagel) dapsone ERY (erythromycin) ERYGEL (erythromycin) erythromycin gel, swabs, solution EVOCLIN (clindamycin) KLARON (sulfacetamide) sulfacetamide RETINOIDS RETIN-A (tretinoin) adapalene tretinoin cream AKLIEF (trifarotene) ALTRENO (tretinoin) ARAZLO (tazarotene) ATRALIN (tretinoin) AVITA (tretinoin) DIFFERIN (adapalene) FABIOR (tazarotene) PLIXDA (adapalene) 1 Drug coverage subject to the rules and regulations set forth in Sec. 1927 of Social Security Act.This is not an all-inclusive list of available covered drugs and includes only managed categories. Unless otherwise stated, the listing of a particular brand or generic name includes all dosage forms of that drug. NR indicates a new drug that has not yet been reviewed by the P&T Committee. PREFERRED BRANDS will not count toward the two brand monthly Rx limit. Drugs highlighted in yellow denote a change in PDL status. An * denotes existing users will be grandfathered; grandfathering is defined as approving a Non-Preferred agent for an existing user; all other changes will not qualify for grandfathering. -

Third Quarter 2017 Update
JULY 2017 THIRD QUARTER 2017 UPDATE CHANGES TO THE HIGHMARK DRUG FORMULARIES Following is the Third Quarter 2017 update to the Highmark Drug Formularies and pharmaceutical management procedures. The formularies and pharmaceutical management procedures are updated on a quarterly basis, and the following changes reflect the decisions made in May 2017 by our Pharmacy and Therapeutics Committee. These updates are effective on the dates noted throughout this document. Please reference the guide below to navigate this communication: Section I. Highmark Commercial and Healthcare Reform Formularies A. Changes to the Highmark Comprehensive Formulary and the Highmark Comprehensive Healthcare Reform Formulary B. Changes to the Highmark Progressive Formulary and the Highmark Progressive Healthcare Reform Formulary C. Changes to the Highmark Healthcare Reform Essential Formulary D. Updates to the Pharmacy Utilization Management Programs 1. Prior Authorization Program 2. Managed Prescription Drug Coverage (MRxC) Program 3. Quantity Level Limit (QLL) Programs Section II. Highmark Medicare Part D Formularies A. Changes to the Highmark Medicare Part D 5-Tier Incentive Formulary B. Changes to the Highmark Medicare Part D 5-Tier Closed Formulary C. Additions to the Specialty Tier D. Updates to the Pharmacy Utilization Management Programs 1. Prior Authorization Program 2. Managed Prescription Drug Coverage (MRxC) Program 3. Quantity Level Limit (QLL) Program As an added convenience, you can also search our drug formularies and view utilization management policies on the Provider Resource Center (accessible via NaviNet® or our website). Click the Pharmacy/Formulary Information link from the menu on the left Highmark Blue Shield is an independent licensee of the Blue Cross and Blue Shield Association. -
January 2020: Additions and Deletions to the Drug Product List
Prescription and Over-the-Counter Drug Product List 40TH EDITION Cumulative Supplement Number 01 : January 2020 ADDITIONS/DELETIONS FOR PRESCRIPTION DRUG PRODUCT LIST ACETAMINOPHEN; HYDROCODONE BITARTRATE TABLET;ORAL HYDROCODONE BITARTRATE AND ACETAMINOPHEN >A> AA XIROMED 325MG;5MG A 211690 001 Feb 07, 2020 Jan NEWA >A> AA 325MG;7.5MG A 211690 002 Feb 07, 2020 Jan NEWA >A> AA 325MG;10MG A 211690 003 Feb 07, 2020 Jan NEWA ACYCLOVIR CREAM;TOPICAL ACYCLOVIR >D> AB PERRIGO UK FINCO 5% A 208702 001 Feb 04, 2019 Jan CHRS >A> AB ! 5% A 208702 001 Feb 04, 2019 Jan CHRS ZOVIRAX >D> AB +! BAUSCH 5% N 021478 001 Dec 30, 2002 Jan CHRS >A> AB + 5% N 021478 001 Dec 30, 2002 Jan CHRS OINTMENT;TOPICAL ACYCLOVIR >A> AB XIROMED 5% A 201501 001 Jan 29, 2020 Jan NEWA TABLET;ORAL ACYCLOVIR >D> AB HETERO LABS LTD V 800MG A 203834 002 Oct 29, 2013 Jan CHRS >A> AB ! 800MG A 203834 002 Oct 29, 2013 Jan CHRS >D> ZOVIRAX >D> AB + MYLAN 400MG N 020089 001 Apr 30, 1991 Jan DISC >A> + @ 400MG N 020089 001 Apr 30, 1991 Jan DISC >D> AB +! 800MG N 020089 002 Apr 30, 1991 Jan DISC >A> + @ 800MG N 020089 002 Apr 30, 1991 Jan DISC AMINO ACIDS; CALCIUM CHLORIDE; DEXTROSE; MAGNESIUM SULFATE; POTASSIUM CHLORIDE; SODIUM ACETATE; SODIUM GLYCEROPHOSPHATE; SOYBEAN OIL EMULSION;INTRAVENOUS PERIKABIVEN IN PLASTIC CONTAINER >D> + FRESENIUS KABI USA 2.4%;20MG/100ML;6.8GM/100ML;68M N 200656 003 Aug 25, 2014 Jan CHRS G/100ML;124MG/100ML;170MG/100ML ;105MG/100ML;3.5GM/100ML (2400ML) >A> +! 2.4%;20MG/100ML;6.8GM/100ML;68M N 200656 003 Aug 25, 2014 Jan CHRS G/100ML;124MG/100ML;170MG/100ML -

Final Belgian List
Final Belgian list: (Last update 07/02/2019) Marketing name Active substance MAH Reason on list ABASAGLAR (ABASRIA previously)INSULINE GLARGINE ELI LILLY REGIONAL OPERATIONS GMBH New Biological ACARIZAX STANDARDIZED ALLERGEN EXTRACT FROM HOUSE DUSTALK-ABELLO MITES A/S New Biological ACCOFIL FILGASTRIM ACCORD HEALTHCARE LIMITED New Biological ADCETRIS BRENTUXIMAB VEDOTINE TAKEDA GLOBAL R&D CENTRE (EUROPE) LTD New AS, Cond Auth, PASS ADEMPAS RIOCIGUAT BAYER PHARMA New AS ADYNOVI RURIOCTOCOG ALFA PEGOL BAXALTA INNOVATIONS GMBH New AS, PASS AIMOVIG ERENUMAB NOVARTIS EUROPHARM LIMITED New AS, New Biological AFSTYLA LONOCTOCOG ALFA CSL BEHRING GmbH New AS STANDARDIZED ALLERGEN EXTRACT FROM HOUSE AITARO ALK-ABELLO A/S New Biological DUST MITES AKYNZEO NETUPITANT, PALONOSETRON HELSINN BIREX PHARMACEUTICALS LTD New AS ALECENSA ALECTINIB ROCHE REGISTRATION LIMITED New AS, Cond Auth ALOFISEL DAVASTROCEL TiGenix SAU New AS ALPHA-RIX TETRA INFLUENZA VACCINE INACTIVATED GLAXOSMITHKLINE BIOLOGICALS S.A. New Biological ALPIVAB PERAMIVIR BIOCRYST UK LTD New AS ALPROLIX EFTRENOCOG ALFA BIOGEN IDEC LTD New AS ALUNBRIG BRIGATINIB TAKEDA PHARMA A/S New AS AMGEVITA ADALIMUMAB AMGEN EUROPE BV New biological STANDARDIZED ALLERGEN EXTRACT FROM HOUSE AMITEND ALK-ABELLO A/S New Biological DUST MITES AMITIZA LUBIPROSTON Sucampo Pharma Europe Ltd New AS ANORO GLAXO GROUP LTD New AS, PASS UMECLIDINIUM BROMIDE, VILANTEROL TRIFENATATE APLEEK ETHINYL ESTRADIOL/GESTODENE BAYER PHARMA AG PASS ATRIANCE NELARABINE 5.00 MG/ML GLAXO GROUP LTD Auth under excep circumstances -

Xermelo (Telotristat Ethyl)
Prior Authorization Criteria Xermelo (telotristat ethyl) Policy Number: C16325-A CRITERIA EFFECTIVE DATES: ORIGINAL EFFECTIVE DATE LAST REVIEWED DATE NEXT REVIEW DUE BY OR BEFORE 7/2019 12/2/2020 1/26/2022 HCPCS CODING TYPE OF CRITERIA LAST P&T APPROVAL/VERSION J8499 (NOC) RxPA Q1 2021 20210127C16325-A PRODUCTS AFFECTED: Xermelo (telotristat ethyl) DRUG CLASS: Tryptophan hydroxylase inhibitor ROUTE OF ADMINISTRATION: oral PLACE OF SERVICE: Retail Pharmacy AVAILABLE DOSAGE FORMS: 250mg tablets FDA-APPROVED USES: for the treatment of carcinoid syndrome diarrhea in combination with somatostatin analog (SSA) therapy in adults inadequately controlled by SSA therapy COMPENDIAL APPROVED OFF-LABELED USES: None COVERAGE CRITERIA: INITIAL AUTHORIZATION DIAGNOSIS: Carcinoid syndrome diarrhea REQUIRED MEDICAL INFORMATION: A. CARCINOID SYNDROME DIARRHEA: 1. Documentation of member has a carcinoid/neuroendocrine tumor and has a diagnosis of carcinoid syndrome AND 2. Member has been receiving therapy with the FDA-approved maximum (or highest tolerated) dose of a somatostatin analog therapy (SSA) (i.e., octreotide solution/depot or lanreotide depot) for at least 3 months AND 3. Member will continue to receive this SSA therapy in combination with Xermelo; AND 4. Member has had an inadequate response to antidiarrheals (e.g., loperamide); Molina Healthcare, Inc. confidential and proprietary © 2021 This document contains confidential and proprietary information of Molina Healthcare and cannot be reproduced, distributed, or printed without written permission -

Antibodies to Watch in 2021 Hélène Kaplona and Janice M
MABS 2021, VOL. 13, NO. 1, e1860476 (34 pages) https://doi.org/10.1080/19420862.2020.1860476 PERSPECTIVE Antibodies to watch in 2021 Hélène Kaplona and Janice M. Reichert b aInstitut De Recherches Internationales Servier, Translational Medicine Department, Suresnes, France; bThe Antibody Society, Inc., Framingham, MA, USA ABSTRACT ARTICLE HISTORY In this 12th annual installment of the Antibodies to Watch article series, we discuss key events in antibody Received 1 December 2020 therapeutics development that occurred in 2020 and forecast events that might occur in 2021. The Accepted 1 December 2020 coronavirus disease 2019 (COVID-19) pandemic posed an array of challenges and opportunities to the KEYWORDS healthcare system in 2020, and it will continue to do so in 2021. Remarkably, by late November 2020, two Antibody therapeutics; anti-SARS-CoV antibody products, bamlanivimab and the casirivimab and imdevimab cocktail, were cancer; COVID-19; Food and authorized for emergency use by the US Food and Drug Administration (FDA) and the repurposed Drug Administration; antibodies levilimab and itolizumab had been registered for emergency use as treatments for COVID-19 European Medicines Agency; in Russia and India, respectively. Despite the pandemic, 10 antibody therapeutics had been granted the immune-mediated disorders; first approval in the US or EU in 2020, as of November, and 2 more (tanezumab and margetuximab) may Sars-CoV-2 be granted approvals in December 2020.* In addition, prolgolimab and olokizumab had been granted first approvals in Russia and cetuximab saratolacan sodium was first approved in Japan. The number of approvals in 2021 may set a record, as marketing applications for 16 investigational antibody therapeutics are already undergoing regulatory review by either the FDA or the European Medicines Agency. -

XERMELO, INN-Telotristat-Epitrate
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 for how to report adverse reactions. 1. NAME OF THE MEDICINAL PRODUCT Xermelo 250 mg film-coated tablets 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each film-coated tablet contains telotristat etiprate equivalent to 250 mg telotristat ethyl. Excipient with known effect Each tablet contains 168 mg of lactose. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Film-coated tablet. White to off-white film-coated oval tablets (approximately 17 mm long by 7.5 mm wide) with ‘T-E’ debossed on one side and ‘250’ debossed on the other side. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Xermelo is indicated for the treatment of carcinoid syndrome diarrhoea in combination with somatostatin analogue (SSA) therapy in adults inadequately controlled by SSA therapy. 4.2 Posology and method of administration Posology The recommended dose is 250 mg three times daily (tid). Available data suggest that clinical response is usually achieved within 12 weeks of treatment. It is recommended to reassess the benefit of continued therapy in a patient not responding within this time period. Based on the high inter-subject variability observed, accumulation in a subset of patients with carcinoid syndrome cannot be excluded. Therefore, intake of higher doses is not recommended (see section 5.2). Missed doses In the event of a missed dose, patients should take their subsequent dose at the next scheduled time point.