Highlighted the Attributes of Dupixent. Syed Mahmod from PYC Therapeutic: Highlighted the Attributes of Emflaza
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) Patent Application Publication (10) Pub. No.: US 2013/0253056A1 Nemas Et Al
US 20130253 056A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0253056A1 Nemas et al. (43) Pub. Date: Sep. 26, 2013 (54) CONTINUOUS ADMINISTRATION OF (60) Provisional application No. 61/179,511, filed on May LEVODOPA AND/OR DOPA 19, 2009. DECARBOXYLASE INHIBITORS AND COMPOSITIONS FOR SAME Publication Classification (71) Applicant: NEURODERM, LTD., Ness-Ziona (IL) (51) Int. Cl. A63L/216 (2006.01) (72) Inventors: Mara Nemas, Gedera (IL); Oron (52) U.S. Cl. Yacoby-Zeevi, Moshav Bitsaron (IL) CPC .................................... A6 IK3I/216 (2013.01) USPC .......................................................... 514/538 (73) Assignee: Neuroderm, Ltd., Ness-Ziona (IL) (57) ABSTRACT (21) Appl. No.: 13/796,232 Disclosed herein are for example, liquid aqueous composi (22) Filed: Mar 12, 2013 tions that include for example an ester or salt of levodopa, or an ester or salt of carbidopa, and methods for treating neuro Related U.S. Application Data logical or movement diseases or disorders such as restless leg (63) Continuation-in-part of application No. 12/961,534, syndrome, Parkinson's disease, secondary parkinsonism, filed on Dec. 7, 2010, which is a continuation of appli Huntington's disease, Parkinson's like syndrome, PSP. MSA, cation No. 12/836,130, filed on Jul. 14, 2010, now Pat. ALS, Shy-Drager syndrome, dystonia, and conditions result No. 7,863.336, which is a continuation of application ing from brain injury including carbon monoxide or manga No. 12/781,357, filed on May 17, 2010, now Pat. No. nese intoxication, using Substantially continuous administra 8,193,243. tion of levodopa and/or carbidopa or ester and/or salt thereof. -

PSP: Some Answers
PSP: Some Answers Lawrence I. Golbe, MD Professor of Neurology, Rutgers Robert Wood Johnson Medical School Director of Clinical Affairs and Scientific Advisory Board Chairman, CurePSP October 2017 What is Progressive Supranuclear Palsy (PSP)? Of the approximately five to seven of every 100,000 people in Canada with progressive supranuclear palsy (PSP), few, if any, had ever heard of the disease before their diagnosis. In fact, most patients with PSP report that their family doctors knew nothing about it until a neurologist made the diagnosis. As of now, three of every four people with a diagnosis of PSP could have been diagnosed earlier, if their doctor had suspected it and performed the appropriate examination. However, it is appearing in medical journals more and more often, which will help doctors become familiar with PSP. This pamphlet should help patients and their families do the same. Why has no one heard of PSP? PSP is rare: no one even realized it existed until 1963, when several patients were first described at a national neurology research convention and the disease was given its name. In retrospect, at least 12 cases of PSP had appeared in the medical literature between 1909 and 1962, but because of its resemblance to Parkinson’s, it wasn’t recognized as a distinct disease. The brain under the microscope is almost identical to that of “post-encephalitic parkinsonism,” a common condition in the early 20th century but now nearly extinct, which also made for erroneous diagnoses during that era. Although PSP is slightly more common than the well-known amyotrophic lateral sclerosis (called ALS, or Lou Gehrig’s disease in the U.S. -

Carbidopa and Levodopa | Memorial Sloan Kettering Cancer Center
PATIENT & CAREGIVER EDUCATION Carbidopa and Levodopa This information from Lexicomp® explains what you need to know about this medication, including what it’s used for, how to take it, its side effects, and when to call your healthcare provider. Brand Names: US Duopa; Rytary; Sinemet; Sinemet CR [DSC] Brand Names: Canada AA-Levocarb CR; APO-Levocarb; APO-Levocarb 100/25; APO-Levocarb 250/25; DOM-Levo-Carbidopa; Duodopa; MINT-Levocarb; PMS-Levocarb CR; Pro-Lecarb; PRO-Levocarb-100/25 [DSC]; Sinemet 100/25; Sinemet 250/25; Sinemet CR 200/50 [DSC]; Sinemet CR [DSC]; TEVA-Levocarbidopa What is this drug used for? It is used to treat Parkinson’s disease. It is used to treat signs like Parkinson’s disease caused by other health problems. It may be given to you for other reasons. Talk with the doctor. What do I need to tell my doctor BEFORE I take this drug? If you are allergic to this drug; any part of this drug; or any other drugs, foods, or substances. Tell your doctor about the allergy and what signs you had. If you have any of these health problems: Glaucoma, a skin lump or growth, or a history of skin cancer. Carbidopa and Levodopa 1/10 If you are taking any of these drugs: Reserpine or tetrabenazine. If you are taking any of these drugs: Linezolid or methylene blue. If you have taken certain drugs for depression or Parkinson’s disease in the last 14 days. This includes isocarboxazid, phenelzine, tranylcypromine, selegiline, or rasagiline. Very high blood pressure may happen. -

Summary of Drug Limitations Mary C
RON DESANTIS GOVERNOR SUMMARY OF DRUG LIMITATIONS MARY C. MAYHEW SECRETARY **Medications listed in this document may or may not require a prior authorization. Please view the Preferred Drug List at: http://ahca.myflorida.com/Medicaid/Prescribed_Drug/pharm_thera/fmpdl.shtml** Summary of Drug Limitations Abilify (aripiprazole) 2mg, 5mg, 20mg, 30mg tablets Minimum age = 6; Maximum of 1 tablet per day Abilify (aripiprazole) 10mg, 15mg tablets Minimum age = 6; Maximum of 15mg per day for ages = 6 - 11; Maximum of 30mg per day for ages = 12-17 Maximum of 1 tablet per day Abilify (aripiprazole) Discmelt 10mg, 15mg tabs Minimum age = 6; Maximum of 15mg per day for ages = 6 - 11; Maximum of 30mg per day for ages = 12-17; Maximum of 2 tablets per day Abilify (aripiprazole) 1mg/ml solution Minimum age = 6; Maximum of 15ml per day for ages = 6 - 11; Maximum of 30ml per day for ages = 12-17; Maximum of 30ml per day for ages =/> 18 Abilify Maintena (aripiprazole) syringe/vial Minimum age = 18; Maximum of 1 syringe or vial every 28 days Absorica (isotretinoin) 10mg, 20mg, 25mg,30mg, 35mg, & Minimum age = 12 40mg capsules Abstral (fentanyl citrate) sublingual tablets Minimum age = 18; Maximum of 4 sublingual tablets per day Acanya (benzoyl peroxide/clindamycin)Gel, gel pump Minimum Age= 12 Accolate (zafirlukast) tablets Maximum of 3 tablets per day Aciphex (rabeprazole) 5mg, 10mg sprinkle capsules Minimum age = 1; Maximum age = 11; Maximum of 1 capsule per day Aciphex (rabeprazole) 20mg tablets Minimum age = 1; Maximum of 2 tablets per day Actemra (tocilizumab) 80mg/4ml, 200mg/10ml, Minimum age= 2 400mg/20ml Vials, & 162mg/0.9ml Syringe Actimmune (Interferon Gamma-1b) Maximum of 6ml every 28days Actiq (fentanyl citrate) Lozenges Minimum age = 18; Maximum of 4 lozenges per day Activella (estradiol/norethindrone) tablets Minimum age = 18 Updated 02/28/2019 1 RON DESANTIS GOVERNOR SUMMARY OF DRUG LIMITATIONS MARY C. -

XERMELO™ (Telotristat Ethyl)
PHARMACY COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 5/18/2017 SECTION: DRUGS LAST REVIEW DATE: 5/20/2021 LAST CRITERIA REVISION DATE: 5/20/2021 ARCHIVE DATE: XERMELO™ (telotristat ethyl) Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Pharmacy Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Pharmacy Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide BCBSAZ complete medical rationale when requesting any exceptions to these guidelines. The section identified as “Description” defines or describes a service, procedure, medical device or drug and is in no way intended as a statement of medical necessity and/or coverage. The section identified as “Criteria” defines criteria to determine whether a service, procedure, medical device or drug is considered medically necessary or experimental or investigational. State or federal mandates, e.g., FEP program, may dictate that any drug, device or biological product approved by the U.S. Food and Drug Administration (FDA) may not be considered experimental or investigational and thus the drug, device or biological product may be assessed only on the basis of medical necessity. Pharmacy Coverage Guidelines are subject to change as new information becomes available. For purposes of this Pharmacy Coverage Guideline, the terms "experimental" and "investigational" are considered to be interchangeable. BLUE CROSS®, BLUE SHIELD® and the Cross and Shield Symbols are registered service marks of the Blue Cross and Blue Shield Association, an association of independent Blue Cross and Blue Shield Plans. -

Azilect, INN-Rasagiline
SCIENTIFIC DISCUSSION 1. Introduction AZILECT is indicated for the treatment of idiopathic Parkinson’s disease (PD) as monotherapy (without levodopa) or as adjunct therapy (with levodopa) in patients with end of dose fluctuations. Rasagiline is administered orally, at a dose of 1 mg once daily with or without levodopa. Parkinson’s disease is a common neurodegenerative disorder typified by loss of dopaminergic neurones from the basal ganglia, and by a characteristic clinical syndrome with cardinal physical signs of resting tremor, bradikinesia and rigidity. The main treatment aims at alleviating symptoms through a balance of anti-cholinergic and dopaminergic drugs. Parkinson’s disease (PD) treatment is complex due to the progressive nature of the disease, and the array of motor and non-motor features combined with early and late side effects associated with therapeutic interventions. Rasagiline is a chemical inhibitor of the enzyme monoamine oxidase (MAO) type B which has a major role in the inactivation of biogenic and diet-derived amines in the central nervous system. MAO has two isozymes (types A and B) and type B is responsible for metabolising dopamine in the central nervous system; as dopamine deficiency is the main contributing factor to the clinical manifestations of Parkinson’s disease, inhibition of MAO-B should tend to restore dopamine levels towards normal values and this improve the condition. Rasagiline was developed for the symptomatic treatment of Parkinson’s disease both as monotherapy in early disease and as adjunct therapy to levodopa + aminoacids decarboxylase inhibitor (LD + ADI) in patients with motor fluctuations. 2. Quality Introduction Drug Substance • Composition AZILECT contains rasagiline mesylate as the active substance. -

Parkinson's Disease Fact Sheet
Parkinson’s Disease Fact Sheet About Parkinson’s Disease Parkinson’s disease is a progressive, incurable neurological disorder associated with a loss of dopamine-generating cells in the brain. It is primarily associated with progressive loss of motor control, but it results in a complex array of symptoms, including many non-motor symptoms. Parkinson’s impacts an estimated one million people in the United States. Critical Clinical Care Considerations • To avoid serious side effects, Parkinson’s patients need their medications on time, every time — do not skip or postpone doses. • Write down the exact times of day medications are to be administered so that doses are given on the same schedule the patient follows at home. • Do not substitute Parkinson’s medications or stop levodopa therapy abruptly. • Resume medications immediately following procedures, unless vomiting or severely incapacitated. • If an antipsychotic is necessary, use pimavanserin (Nuplazid), quetiapine (Seroquel) or clozapine (Clozaril). • Be alert for symptoms of dysphagia (trouble swallowing) and risk of pneumonia. • Ambulate as soon as medically safe. Patients may require assistance. Common Symptoms of Parkinson’s Disease Motor Non-Motor • Shaking or tremor at rest • Depression • Bradykinesia or freezing (being stuck • Anxiety in place when attempting to walk) • Constipation • Low voice volume or muffled speech • Cognitive decline and dementia • Lack of facial expression • Impulse control disorders • Stiffness or rigidity of the arms, legs • Orthostatic hypotension or -

New Drugs Approved in FY 2014
New Drugs Approved in FY 2014 New Active Ingredient(s) Review Brand Name Approval/ Approval Date No. (underlined: new active Notes Category (Applicant Company) Partial ingredient) Change 1 Jul. 4, 2014 1 Dovobet Ointment Approval (1) Calcipotriol A new combination drug indicated for the treatment (Leo Pharma K.K.) hydrate/ of psoriasis vulgaris. (2) Betamethasone dipropionate 1 Aug. 29, 2014 2 Rituxan Injection 10 mg/mL Change Rituximab (genetical A drug with a new additional indication and a new (Zenyaku Kogyo Co., Ltd.) recombination) dosage for the treatment of refractory nephrotic syndrome (for use in patients with frequent recurrence or steroid-dependent). [Orphan drug] 1 Sep. 19, 2014 3 Thymoglobuline for Intravenous Infusion 25 mg Change Anti-human thymocyte A drug with a new additional indication and a new (Sanofi K.K.) immunoglobulin, rabbit dosage for the treatment of acute rejection after the liver, heart, lungs, pancreas, or small intestinal transplantation. 1 Sep. 26, 2014 4 Fomepizole Intravenous Infusion 1.5 g "Takeda" Approval Fomepizole A drug with a new active ingredient indicated for the (Takeda Pharmaceutical Company Limited) treatment of ethylene glycol and methanol poisonings. 1 Dec. 26, 2014 5 Pariet Tablets 5 mg Approval Rabeprazole sodium A drug with a new additional indication and a new Pariet Tablets 10 mg Change dosage in a newly-added dosage form, and a drug (Eisai Co., Ltd.) with a new additional indication and a new dosage for the prevention of recurrence of gastric ulcer or duodenal ulcer in patients treated -

S41467-019-09610-2.Pdf
Corrected: Author correction ARTICLE https://doi.org/10.1038/s41467-019-09610-2 OPEN Mechanism-based tuning of insect 3,4-dihydroxyphenylacetaldehyde synthase for synthetic bioproduction of benzylisoquinoline alkaloids Christopher J. Vavricka1, Takanobu Yoshida1, Yuki Kuriya1, Shunsuke Takahashi 1, Teppei Ogawa2, Fumie Ono3, Kazuko Agari1, Hiromasa Kiyota4, Jianyong Li5, Jun Ishii 1, Kenji Tsuge1, Hiromichi Minami6, Michihiro Araki1,3, Tomohisa Hasunuma1,7 & Akihiko Kondo 1,7,8 1234567890():,; Previous studies have utilized monoamine oxidase (MAO) and L-3,4-dihydroxyphenylalanine decarboxylase (DDC) for microbe-based production of tetrahydropapaveroline (THP), a benzylisoquinoline alkaloid (BIA) precursor to opioid analgesics. In the current study, a phylogenetically distinct Bombyx mori 3,4-dihydroxyphenylacetaldehyde synthase (DHPAAS) is identified to bypass MAO and DDC for direct production of 3,4-dihydrox- yphenylacetaldehyde (DHPAA) from L-3,4-dihydroxyphenylalanine (L-DOPA). Structure- based enzyme engineering of DHPAAS results in bifunctional switching between aldehyde synthase and decarboxylase activities. Output of dopamine and DHPAA products is fine- tuned by engineered DHPAAS variants with Phe79Tyr, Tyr80Phe and Asn192His catalytic substitutions. Balance of dopamine and DHPAA products enables improved THP biosynthesis via a symmetrical pathway in Escherichia coli. Rationally engineered insect DHPAAS produces (R,S)-THP in a single enzyme system directly from L-DOPA both in vitro and in vivo, at higher yields than that of the wild-type enzyme. However, DHPAAS-mediated downstream BIA production requires further improvement. 1 Graduate School of Science, Technology and Innovation, Kobe University, 1-1 Rokkodai-cho, Nada-ku, Kobe 657-8501, Japan. 2 Mitsui Knowledge Industry Co., Ltd. (MKI), 2-3-33 Nakanoshima, Kita-ku, Osaka 530-0005, Japan. -

Parkinson's and Diet
MICHAELJFOX.ORG Parkinson’s and Diet: Overview No one specific diet is recommended for with aging and Parkinson’s disease. A diet everyone with Parkinson’s disease (PD). Still, high in antioxidants may therefore help to what you eat may impact how well your offset oxidative stress, although proof is medication works and can help some of the lacking at this time. non-motor symptoms that are associated with PD. For general health and well-being, doctors encourage people with Parkinson’s SHOPPING LIST FOR FOODS to follow a balanced diet, which includes whole grains, healthy fats (in foods like nuts, CONTAINING ANTIOXIDANTS avocado and olive oil) and antioxidants. Vegetables: artichokes, okra, kale, bell peppers, potatoes Antioxidants are molecules that clear out Fruits: berries, pears, apples, grapes free radicals — substances that are potentially toxic to cells. Free radicals are Legumes: kidney beans, edamame, lentils formed in the body from normal metabolism, Nuts: pecans, walnuts, hazelnuts but are increased by exposure to environmental Dark chocolate toxins, such as cigarette smoke and air Red wine (in moderation), coffee, tea pollution. Free radicals contribute to a condition called oxidative stress, which is associated The Michael J. Fox Foundation for Parkinson's Research | A Practical Guide on Parkinson’s Disease and Diet 2 Diet and Parkinson’s Medications Parkinson’s and Constipation If you take certain Parkinson’s medications, dietary adjust- Constipation is, unfortunately, common among people with ments may help the drugs work more effectively or avoid side Parkinson’s disease. Not only is this non-motor symptom effects. uncomfortable, it can interfere with medication absorption Carbidopa/levodopa — contained in Sinemet, Sinemet CR, and effectiveness. -
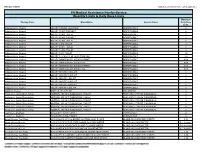
Quantity Limits/Daily Dose Limits
Effective 07/20/21 Alphabetical by Brand Name (when applicable) PA Medical Assistance Fee-for-Service Quantity Limits & Daily Dose Limits Maximum Therapy Class Brand Name Generic Name Daily Dose Limit Antipsychotics, Atypical ABILIFY 1 MG/ML SOLUTION ARIPIPRAZOLE 25 Antipsychotics, Atypical ABILIFY 10 MG DISCMELT ARIPIPRAZOLE 2 Antipsychotics, Atypical ABILIFY 10 MG TABLET ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY 15 MG DISCMELT ARIPIPRAZOLE 2 Antipsychotics, Atypical ABILIFY 15 MG TABLET ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY 2 MG TABLET ARIPIPRAZOLE 2 Antipsychotics, Atypical ABILIFY 20 MG TABLET ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY 30 MG TABLET ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY 5 MG TABLET ARIPIPRAZOLE 1.5 Antipsychotics, Atypical ABILIFY 9.75 MG/1.3 ML INJECTION VIAL ARIPIPRAZOLE 3.9 Antipsychotics, Atypical ABILIFY MAINTENA ER 300 MG SYRINGE ARIPIPRAZOLE 0.04 Antipsychotics, Atypical ABILIFY MAINTENA ER 300 MG VIAL ARIPIPRAZOLE 0.04 Antipsychotics, Atypical ABILIFY MAINTENA ER 400 MG SYRINGE ARIPIPRAZOLE 0.04 Antipsychotics, Atypical ABILIFY MAINTENA ER 400 MG VIAL ARIPIPRAZOLE 0.04 Antipsychotics, Atypical ABILIFY MYCITE 10 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 15 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 2 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 20 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 30 MG KIT ARIPIPRAZOLE 1 Antipsychotics, Atypical ABILIFY MYCITE 5 MG KIT ARIPIPRAZOLE 1 Antivirals, Herpes ABREVA 10% -

ZELAPAR™ (Selegiline Hydrochloride) Orally Disintegrating Tablets
ZELAPAR™ (selegiline hydrochloride) orally disintegrating tablets DESCRIPTION ZELAPARTM Orally Disintegrating Tablets contain selegiline hydrochloride, a levorotatory acetylenic derivative of phenthylamine. Selegiline hydrochloride is described chemically as: (-)-(R)-N, α-dimethyl-N-2-propynylphenethylamine hydrochloride and its structural formula is: Its empirical formula is C13H17N·HCl, representing a molecular weight of 223.75. Selegiline hydrochloride is a white to almost white crystalline powder that is freely soluble in water, chloroform, and methanol. ZELAPARTM Orally Disintegrating Tablets are available for oral administration (not to be swallowed) in a strength of 1.25 mg. Each lyophilized orally disintegrating tablet contains the following inactive ingredients: gelatin, mannitol, glycine, aspartame, citric acid, yellow iron oxide, and grapefruit flavor. CLINICAL PHARMACOLOGY The mechanisms accounting for selegiline’s beneficial adjunctive action in the treatment of Parkinson’s disease are not fully understood. Inhibition of monoamine oxidase type B (MAO-B) activity is generally considered to be of primary importance; in addition, there is evidence that selegiline may act through other mechanisms to increase dopaminergic activity. Selegiline is best known as an irreversible inhibitor of monoamine oxidase (MAO), an intracellular enzyme associated with the outer membrane of mitochondria. Selegiline inhibits MAO by acting as a suicide substrate for the enzyme; that is, it is converted by MAO to an active moiety which combines irreversibly with the active site and/or the enzyme’s essential flavin adenine dinucleotide (FAD) cofactor. Because selegiline has greater affinity for type B rather than for type A active sites, it can serve as a selective inhibitor of MAO type B if it is administered at the recommended dose.