Dissertation Extrinsic and Intrinsic
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
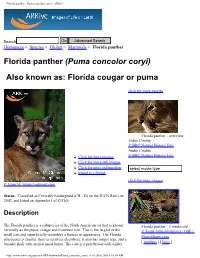
Florida Panther - Puma Concolor Coryi - Arkive
Florida panther - Puma concolor coryi - ARKive Search Homepage > Species > Global > Mammals > Florida panther Florida panther (Puma concolor coryi) Also known as: Florida cougar or puma click for more movies Florida panther - overview Video Credits: © BBC Natural History Unit Audio Credits: © BBC Natural History Unit ● Click for more movies ● Click for more still images ● Click for more information ● Email to a friend click for more images © Lynn M. Stone / naturepl.com Status: Classified as Critically Endangered (CR - D) on the IUCN Red List 2002, and listed on Appendix I of CITES. Description The Florida panther is a subspecies of the North American cat that is known Florida panther - 3 weeks old variously as the puma, cougar and mountain lion. This is the largest of the © Frank Schneidermeyer / OSF / small cats and superficially resembles a lioness in appearance. The Florida Photolibrary.com subspecies is smaller than its relatives elsewhere; it also has longer legs, and a [ medium ] [ large ] broader skull with arched nasal bones. The coat is a pale brown with whiter http://www.arkive.org/species/GES/mammals/Puma_concolor_coryi/ (1 of 2)4/6/2005 8:16:04 AM Florida panther - Puma concolor coryi - ARKive underparts and a black tip at the end of the long tail. Infants have a spotted coat and blue eyes. Florida panthers often have crooked ends to their tails, and whorls of hair on their backs; these are thought not to be characteristic of the subspecies however, and may be signs of inbreeding. Click for more information Florida panther - 5 months old © Bob Bennett / OSF / Photolibrary.com [ medium ] [ large ] © Wildscreen 2004 By using this website you agree to the Terms of Use About ARKive | Competition | Contact | Newsletter | FAQ | Links http://www.arkive.org/species/GES/mammals/Puma_concolor_coryi/ (2 of 2)4/6/2005 8:16:04 AM. -

Common Modular Structure of Lentivirus Ltrs
VIROLOGY 224, 256–267 (1996) ARTICLE NO. 0527 Common Modular Structure of Lentivirus LTRs View metadata, citation and similar papers at core.ac.uk brought to you by CORE KORNELIE FRECH,* RUTH BRACK-WERNER,*,† and THOMAS WERNER*,1 provided by Elsevier - Publisher Connector *Institut fu¨rSa¨ugetiergenetik and †Institut fu¨r Molekulare Virologie, GSF-Forschungszentrum fu¨r Umwelt und Gesundheit GmbH, Ingolsta¨dter Landstrasse 1, D-85758 Oberschleißheim, Germany Received April 17, 1996; accepted August 5, 1996 Retroviruses are expressed under the control of viral control regions designated long terminal repeats (LTRs), which contain all signals for transcriptional initiation as well as transcriptional termination. However, retroviral LTRs from different species within a common genus, such as Lentivirus, do not show significant overall sequence homology. We compiled a model of the functional organization of 20 Lentivirus LTRs which we show to recognize all known Lentivirus LTRs. To this end we combined our previously published methods for identification of transcription elements with secondary structure element analysis in a novel modular approach. We deduced descriptions for three new Lentivirus-specific sequence elements present in most of the Lentivirus LTRs but absent in LTRs of other retrovirus families (B, C, D-type, BLV-HTLV, Spuma). Four of the 10 elements defined in our study were primate-specific. We were able to deduce a phylogeny based on our model which agrees in general with the phylogeny derived from the polymerase genes of these viruses. Our model indicated that more than 100 LTRs from the databases are of Lentivirus origin and can be clearly separated from all other LTR types (B, C, D, BLV-HTLV, Spuma). -

Emerging Viruses in the Felidae: Shifting Paradigms
Viruses 2012, 4, 236-257; doi:10.3390/v4020236 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Review Emerging Viruses in the Felidae: Shifting Paradigms Stephen J. O’Brien 1,*,†, Jennifer L. Troyer 2, Meredith A. Brown 3, Warren E. Johnson 1, Agostinho Antunes 4, Melody E. Roelke 2 and Jill Pecon-Slattery 1 1 Laboratory of Genomic Diversity, National Cancer Institute-Frederick, Frederick, MD 21702, USA; E-Mails: [email protected] (W.E.J.); [email protected] (J.P.-S.) 2 SAIC-Frederick, Inc., National Cancer Institute-Frederick, Frederick, MD 21702, USA; E-Mails: [email protected] (J.L.T.); [email protected] (M.E.R.) 3 Banfield Pet Hospital, 800 NE Tillamook Street, Portland, OR 97213, USA; E-Mail: [email protected] 4 CIMAR/CIIMAR, University of Porto, Rua dos Bragas, 177, Porto 4050-123, Portugal; E-Mail: [email protected] † Present Address: Theodosius Dobzhansky Center for Genome Bioinformatics, St. Petersburg University, St. Petersburg, 190000, Russia. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-240-446-1021; Fax: +1-301-662-1413. Received: 1 December 2011; in revised form: 21 December 2011 / Accepted: 11 January 2012 / Published: 7 February 2012 Abstract: The domestic cat is afflicted with multiple viruses that serve as powerful models for human disease including cancers, SARS and HIV/AIDS. Cat viruses that cause these diseases have been studied for decades revealing detailed insight concerning transmission, virulence, origins and pathogenesis. Here we review recent genetic advances that have questioned traditional wisdom regarding the origins of virulent Feline infectious peritonitis (FIP) diseases, the pathogenic potential of Feline Immunodeficiency Virus (FIV) in wild non-domestic Felidae species, and the restriction of Feline Leukemia Virus (FeLV) mediated immune impairment to domestic cats rather than other Felidae species. -

The Florida Panther: a Story of Conflict, Connections and Coexistence
The Florida Panther: A Story of Conflict, Connections and Coexistence Laurie Macdonald Florida Director DefendersDefenders of of Wildlife Wildlife Endangered – US Endangered Species Act of 1973 Population estimate in 1970s = 12-20 Genetic reinvigoration program 1995 Population estimate today = 100-180 Defenders of Wildlife Florida’s panther story Figure prepared by the Florida Fish & Wildlife Conservation Commission Once upon a time…. Sustainable coexistence? Little or no management Abundant Over- Conflict exploited management Photo Credit: Cory, Charles B. Hunting and Fishing in Florida. New York: Arno Press 1970 Rare More numerous Recovery Rebounding management Florida now and future… • 19 million people • 34.7 million acres 3 Foot Sea Level Rise Defenders of Wildlife Chances of successful recovery will be the greatest if the panther is able to travel north on its own and resettle its historic home. Defenders of Wildlife * Core habitat * Connectivity * Coexistence Defenders of Wildlife Conservation of Core and Corridor Habitat Habitat Protection at the Landscape Level Defenders of Wildlife Defenders of Wildlife Amendment 1 – Florida’s Water and Land Legacy State Constitutional Amendment Vote November 4th, 2014 Greater Everglades Refuge Planning Areas Connectivity: Corridors, Linkages, Networks Defenders of Wildlife Defenders of Wildlife ©Mark Lotz, Florida Fish and Wildlife Conservation Commission Defenders of Wildlife Florida Black Bear (Ursus americanus floridanus) American Alligator (Alligator mississippiensis) Defenders of Wildlife -

Evolution of Puma Lentivirus in Bobcats (Lynx Rufus) and Mountain Lions (Puma Concolor) in North America Justin S
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln USDA National Wildlife Research Center - Staff U.S. Department of Agriculture: Animal and Plant Publications Health Inspection Service 2014 Evolution of Puma Lentivirus in Bobcats (Lynx rufus) and Mountain Lions (Puma concolor) in North America Justin S. Lee Colorado State University, [email protected] Sarah N. Bevins USDA National Wildlife Research Center, [email protected] Laurel E. K. Serleys University of California, Los Angeles, [email protected] Winston Vickers University of California, Davis, [email protected] Ken A. Logan Colorado Parks and Wildlife, [email protected] See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/icwdm_usdanwrc Part of the Life Sciences Commons Lee, Justin S.; Bevins, Sarah N.; Serleys, Laurel E. K.; Vickers, Winston; Logan, Ken A.; Aldredge, Mat; Boydston, Erin E.; Lyren, Lisa M.; McBride, Roy; Roelke-Parker, Melody; Pecon-Slattery, Jill; Troyer, Jennifer L.; Riley, Seth P.; Boyce, Walter M.; Crooks, Kevin R.; and VandeWoude, Sue, "Evolution of Puma Lentivirus in Bobcats (Lynx rufus) and Mountain Lions (Puma concolor) in North America" (2014). USDA National Wildlife Research Center - Staff Publications. 1534. https://digitalcommons.unl.edu/icwdm_usdanwrc/1534 This Article is brought to you for free and open access by the U.S. Department of Agriculture: Animal and Plant Health Inspection Service at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in USDA National Wildlife Research Center - Staff ubP lications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Justin S. Lee, Sarah N. -

Florida Panthers Typically Will a Confrontation.Avoid Give Them Escape
You live in Florida panther country If you see a Florida panther A guide to living with Florida panthers are reclusive and rarely seen by people. The Florida panther moves primarily at night. The They normally live in remote, undeveloped areas. chances of seeing a panther are slim. But if you live in printed on recycled paper MyFWC.com/Panther Tallahassee,32399-1600 FL 620 S. MeridianStreet Conservation Commission Florida Fish and Wildlife FishFlorida Wildlife and However, as the number of people in southern Florida Florida panther country, you need to know what to do if grows, there is an increased chance of an encounter you see one. Florida with a Florida panther. Keep children within sight and close to you. Pick This brochure contains some guidelines to help you live up any small children so they don’t panic and run. Panthers safely in Florida panther country. Try to do this without bending over or turning away from the Florida panther. Give them space. Florida panthers typically will avoid a confrontation. Give them a way to escape. Do not run. Running may stimulate a panther’s instinct to chase. Stand and face the animal. Make eye contact to let the panther know you are aware of its presence. Avoid crouching or bending over. Squatting or bending makes you look smaller, resembling a prey- sized animal. Appear larger. Make gestures that indicate you are not prey and that you may be a danger to the panther. Raise your arms. Open your jacket. Throw stones, branches or whatever you can reach without crouching or turning your back. -

Revealed Via Genomic Assessment of Felid Cansines
Evolutionary and Functional Impacts of Short Interspersed Nuclear Elements (SINEs) Revealed via Genomic Assessment of Felid CanSINEs By Kathryn B. Walters-Conte B. S., May 2000, University of Maryland, College Park M. S., May 2002, The George Washington University A Dissertation Submitted to The Faculty of Columbian College of Arts and Sciences of The George Washington University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy May 15 th , 2011 Dissertation Directed By Diana L.E. Johnson Associate Professor of Biology Jill Pecon-Slattery Staff Scientist, National Cancer Institute . The Columbian College of Arts and Sciences of The George Washington University certifies that Kathryn Walters-Conte has passed the Final Examination for the degree of Doctor of Philosophy as of March 24 th , 2011. This is the final and approved form of the dissertation. Evolutionary and Functional Impacts of Short Interspersed Nuclear Elements (SINEs) Revealed via Genomic Assessment of Felid CanSINEs Kathryn Walters-Conte Dissertation Research Committee: Diana L.E. Johnson, Associate Professor of Biology, Dissertation Co-Director Jill Pecon-Slattery, Staff Scientist, National Cancer Institute, Dissertation Co-Director Diana Lipscomb, Ronald Weintraub Chair and Professor, Committee Member Marc W. Allard, Research Microbiologist, U.S. Food and Drug Administration, Committee Member ii Acknowledgements I would like to first thank my advisor and collaborator, Dr. Jill Pecon-Slattery, at the National Cancer Institute of the National Institutes of Health, for generously permitting me to join her research group. Without her mentorship this dissertation would never have been possible. I would also like to express gratitude to my advisor at the George Washington University, Dr. -

Extended and Continuous Decline in Effective Population Size Results in Low Genomic Diversity in the World’S Rarest Hyena Species, the Brown Hyena Michael V
Extended and Continuous Decline in Effective Population Size Results in Low Genomic Diversity in the World’s Rarest Hyena Species, the Brown Hyena Michael V. Westbury,*,1,2 Stefanie Hartmann,2 Axel Barlow,2 Ingrid Wiesel,3,4 Viyanna Leo,5 Rebecca 6,7 6 8 9 10 2 Welch, Daniel M. Parker, Florian Sicks, Arne Ludwig, Love Dalen, and Michael Hofreiter Downloaded from https://academic.oup.com/mbe/article-abstract/35/5/1225/4924857 by Swedish Museum of Natural History user on 14 December 2018 1Natural History Museum of Denmark, University of Copenhagen, Copenhagen, Denmark 2Evolutionary Adaptive Genomics, Institute of Biochemistry and Biology, University of Potsdam, Potsdam, Germany 3Brown Hyena Research Project Trust Fund, Lu¨deritz, Namibia 4Mammal Research Institute, University of Pretoria, Pretoria, South Africa 5Center of Ecosystem Science, University of New South Wales, NSW, Australia 6Wildlife and Reserve Management Research Group, Department of Zoology and Entomology, Rhodes University, Grahamstown, South Africa 7Department of Zoology and Entomology, University of the Free State, Phuthaditjhaba, South Africa 8Tierpark Berlin, Berlin, Germany 9Department of Evolutionary Genetics, Leibniz-Institute for Zoo and Wildlife Research, Berlin, Germany 10Department of Bioinformatics and Genetics, Swedish Museum of Natural History, Stockholm, Sweden *Corresponding author: E-mail: [email protected]. Associate editor: Beth Shapiro Brown hyena raw sequencing reads can be found under the accession codes SAMN07431150–SAMN07431164. The striped hyena assembly can be found at PEQU00000000 and the raw reads can be found at SAMN07212965. The brown hyena mitochondrial genomes can be found at MF593938–MF593952. Abstract Hyenas (family Hyaenidae), as the sister group to cats (family Felidae), represent a deeply diverging branch within the cat-like carnivores (Feliformia). -

The Role of Big Cypress National Preserve (Big Cypress) in Florida Panther Recovery Has Evolved As Research Has Replaced Specula
FLORIDA PANTHER (Puma concolor coryi) RESEARCH AND MONITORING IN BIG CYPRESS NATIONAL PRESERVE 2009-2010 ANNUAL REPORT Photo of FP175 by Ralph Arwood Submitted to U. S. Fish and Wildlife Service (Endangered Species Permit TE146761-1 and Florida Fish and Wildlife Conservation Commission (Special Purpose Permit WX08654) December 31, 2010 Prepared by National Park Service staff Deborah Jansen, Project Leader John Kellam, Biological Technician Annette Johnson, Biological Technician Table of Contents Abstract .................................................................................................................................................... 1 Report Background ................................................................................................................................... 2 Statement of Purpose ................................................................................................................................ 2 Project Goals ............................................................................................................................................ 3 Study Area ............................................................................................................................................... 4 Methods ................................................................................................................................................... 5 Study Area Sampling ........................................................................................................................ -

Florida Panther Puma Concolor Coryi
Florida Panther Puma concolor coryi he Florida panther, a subspecies of mountain lion, is Federal Status: Endangered (March 11, 1967) one of the most endangered large mammals in the Critical Habitat: None Designated Tworld. It is also Floridas state animal. A small Florida Status: Endangered population in South Florida,estimated to number between 30 and 50 adults (30 to 80 total individuals), represents the only Recovery Plan Status: Contribution (May 1999) known remaining wild population of an animal that once Geographic Coverage: South Florida ranged throughout most of the southeastern United States from Arkansas and Louisiana eastward across Mississippi, Alabama, Georgia, Florida and parts of South Carolina and Figure 1. County distribution of the Florida panther since 1981, based on radiotelemetry data. Tennessee. The panther presently occupies one of the least developed areas in the eastern United States; a contiguous system of large private ranches and public conservation lands in Broward, Collier, Glades, Hendry, Lee, Miami-Dade, Monroe, and Palm Beach counties totaling more than 809,400 ha. Geographic isolation, habitat loss, population decline, and associated inbreeding have resulted in a significant loss of genetic variability and overall health of the Florida panther population. Natural gene exchange ceased when the panther became geographically isolated from other subspecies of Puma concolor about a century ago. Population viability projections have concluded that, under current demographic and genetic conditions, the panther would probably become extinct within two to four decades. A genetic management program was implemented with the release of eight female Texas cougars (Puma concolor stanleyana) into South Florida in 1995 (refer to the Management section for a discussion of this program). -

Animal Models for HIV AIDS: a Comparative Review
Comparative Medicine Vol 57, No 1 Copyright 2007 February 2007 by the American Association for Laboratory Animal Science Pages 33-43 Animal Models for HIV AIDS: A Comparative Review Debora S Stump and Sue VandeWoude* Human immunodeficiency virus (HIV), the causative agent for acquired immune deficiency syndrome, was described over 25 y ago. Since that time, much progress has been made in characterizing the pathogenesis, etiology, transmission, and disease syndromes resulting from this devastating pathogen. However, despite decades of study by many investigators, basic questions about HIV biology still remain, and an effective prophylactic vaccine has not been developed. This review provides an overview of the viruses related to HIV that have been used in experimental animal models to improve our knowledge of lentiviral disease. Viruses discussed are grouped as causing (1) nonlentiviral immunodeficiency-inducing diseases, (2) naturally occurring pathogenic infections, (3) experimentally induced lentiviral infections, and (4) nonpathogenic lentiviral infections. Each of these model types has provided unique contributions to our understanding of HIV disease; further, a comparative overview of these models both reinforces the unique attributes of each agent and provides a basis for describing elements of lentiviral disease that are similar across mammalian species. Abbreviations: AIDS, acquired immune deficiency syndrome; BIV, bovine immunodeficiency virus; CAEV, caprine arthritis-encephalitis virus; CRPRC, California Regional Primate Research -

Prior Puma Lentivirus Infection Modifies Early Immune
viruses Article Prior Puma Lentivirus Infection Modifies Early Immune Responses and Attenuates Feline Immunodeficiency Virus Infection in Cats Wendy S. Sprague 1,2,*, Ryan M. Troyer 1,3, Xin Zheng 1, Britta A. Wood 1,4, Martha Macmillian 1, Scott Carver 1,5 ID and Susan VandeWoude 1 1 Department of Molecular Biology, Immunology and Pathology, College of Veterinary Medicine and Biological Sciences, Colorado State University, Fort Collins, CO 80523, USA; [email protected] (R.M.T.); [email protected] (X.Z.); [email protected] (B.A.W.); [email protected] (M.M.); [email protected] (S.C.); [email protected] (S.V.) 2 Sprague Medical and Scientific Communications, LLC, Fort Collins, CO 80528, USA 3 Department of Microbiology and Immunology, University of Western Ontario, London, ON N6A5C1, Canada 4 The Pirbright Institute, Pirbright, Surrey GU24 0NF, UK 5 School of Natural Sciences, University of Tasmania, Hobart, Tasmania 7001, Australia * Correspondence: [email protected]; Tel.: +1-970-223-0333 Received: 22 March 2018; Accepted: 12 April 2018; Published: 20 April 2018 Abstract: We previously showed that cats that were infected with non-pathogenic Puma lentivirus (PLV) and then infected with pathogenic feline immunodeficiency virus (FIV) (co-infection with the host adapted/pathogenic virus) had delayed FIV proviral and RNA viral loads in blood, with viral set-points that were lower than cats infected solely with FIV. This difference was associated with global CD4+ T cell preservation, greater interferon gamma (IFN-γ) mRNA expression, and no cytotoxic T lymphocyte responses in co-infected cats relative to cats with a single FIV infection.