Revealed Via Genomic Assessment of Felid Cansines
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Conservation of the Wildcat (Felis Silvestris) in Scotland: Review of the Conservation Status and Assessment of Conservation Activities
Conservation of the wildcat (Felis silvestris) in Scotland: Review of the conservation status and assessment of conservation activities Urs Breitenmoser, Tabea Lanz and Christine Breitenmoser-Würsten February 2019 Wildcat in Scotland – Review of Conservation Status and Activities 2 Cover photo: Wildcat (Felis silvestris) male meets domestic cat female, © L. Geslin. In spring 2018, the Scottish Wildcat Conservation Action Plan Steering Group commissioned the IUCN SSC Cat Specialist Group to review the conservation status of the wildcat in Scotland and the implementation of conservation activities so far. The review was done based on the scientific literature and available reports. The designation of the geographical entities in this report, and the representation of the material, do not imply the expression of any opinion whatsoever on the part of the IUCN concerning the legal status of any country, territory, or area, or its authorities, or concerning the delimitation of its frontiers or boundaries. The SWCAP Steering Group contact point is Martin Gaywood ([email protected]). Wildcat in Scotland – Review of Conservation Status and Activities 3 List of Content Abbreviations and Acronyms 4 Summary 5 1. Introduction 7 2. History and present status of the wildcat in Scotland – an overview 2.1. History of the wildcat in Great Britain 8 2.2. Present status of the wildcat in Scotland 10 2.3. Threats 13 2.4. Legal status and listing 16 2.5. Characteristics of the Scottish Wildcat 17 2.6. Phylogenetic and taxonomic characteristics 20 3. Recent conservation initiatives and projects 3.1. Conservation planning and initial projects 24 3.2. Scottish Wildcat Action 28 3.3. -

The Taxonomic Status of Badgers (Mammalia, Mustelidae) from Southwest Asia Based on Cranial Morphometrics, with the Redescription of Meles Canescens
Zootaxa 3681 (1): 044–058 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2013 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3681.1.2 http://zoobank.org/urn:lsid:zoobank.org:pub:035D976E-D497-4708-B001-9F8DC03816EE The taxonomic status of badgers (Mammalia, Mustelidae) from Southwest Asia based on cranial morphometrics, with the redescription of Meles canescens ALEXEI V. ABRAMOV1 & ANDREY YU. PUZACHENKO2 1Zoological Institute, Russian Academy of Sciences, Universitetskaya nab. 1, 199034 St. Petersburg, Russia. E-mail: [email protected] 2Institute of Geography, Russian Academy of Sciences, Staromonetnyi per. 22, 109017 Moscow, Russia. E-mail: [email protected] Abstract The Eurasian badgers (Meles spp.) are widespread in the Palaearctic Region, occurring from the British Islands in the west to the Japanese Islands in the east, including the Scandinavia, Southwest Asia and southern China. The morphometric vari- ation in 30 cranial characters of 692 skulls of Meles from across the Palaearctic was here analyzed. This craniometric anal- ysis revealed a significant difference between the European and Asian badger phylogenetic lineages, which can be further split in two pairs of taxa: meles – canescens and leucurus – anakuma. Overall, European badger populations are very sim- ilar morphologically, particularly with regards to the skull shape, but differ notably from those from Asia Minor, the Mid- dle East and Transcaucasia. Based on the current survey of badger specimens available in main world museums, we have recognized four distinctive, parapatric species: Meles meles, found in most of Europe; Meles leucurus from continental Asia; M. -
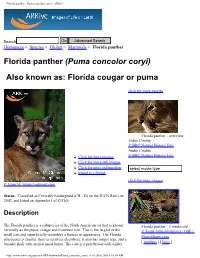
Florida Panther - Puma Concolor Coryi - Arkive
Florida panther - Puma concolor coryi - ARKive Search Homepage > Species > Global > Mammals > Florida panther Florida panther (Puma concolor coryi) Also known as: Florida cougar or puma click for more movies Florida panther - overview Video Credits: © BBC Natural History Unit Audio Credits: © BBC Natural History Unit ● Click for more movies ● Click for more still images ● Click for more information ● Email to a friend click for more images © Lynn M. Stone / naturepl.com Status: Classified as Critically Endangered (CR - D) on the IUCN Red List 2002, and listed on Appendix I of CITES. Description The Florida panther is a subspecies of the North American cat that is known Florida panther - 3 weeks old variously as the puma, cougar and mountain lion. This is the largest of the © Frank Schneidermeyer / OSF / small cats and superficially resembles a lioness in appearance. The Florida Photolibrary.com subspecies is smaller than its relatives elsewhere; it also has longer legs, and a [ medium ] [ large ] broader skull with arched nasal bones. The coat is a pale brown with whiter http://www.arkive.org/species/GES/mammals/Puma_concolor_coryi/ (1 of 2)4/6/2005 8:16:04 AM Florida panther - Puma concolor coryi - ARKive underparts and a black tip at the end of the long tail. Infants have a spotted coat and blue eyes. Florida panthers often have crooked ends to their tails, and whorls of hair on their backs; these are thought not to be characteristic of the subspecies however, and may be signs of inbreeding. Click for more information Florida panther - 5 months old © Bob Bennett / OSF / Photolibrary.com [ medium ] [ large ] © Wildscreen 2004 By using this website you agree to the Terms of Use About ARKive | Competition | Contact | Newsletter | FAQ | Links http://www.arkive.org/species/GES/mammals/Puma_concolor_coryi/ (2 of 2)4/6/2005 8:16:04 AM. -

Felis Silvestris, Wild Cat
The IUCN Red List of Threatened Species™ ISSN 2307-8235 (online) IUCN 2008: T60354712A50652361 Felis silvestris, Wild Cat Assessment by: Yamaguchi, N., Kitchener, A., Driscoll, C. & Nussberger, B. View on www.iucnredlist.org Citation: Yamaguchi, N., Kitchener, A., Driscoll, C. & Nussberger, B. 2015. Felis silvestris. The IUCN Red List of Threatened Species 2015: e.T60354712A50652361. http://dx.doi.org/10.2305/IUCN.UK.2015-2.RLTS.T60354712A50652361.en Copyright: © 2015 International Union for Conservation of Nature and Natural Resources Reproduction of this publication for educational or other non-commercial purposes is authorized without prior written permission from the copyright holder provided the source is fully acknowledged. Reproduction of this publication for resale, reposting or other commercial purposes is prohibited without prior written permission from the copyright holder. For further details see Terms of Use. The IUCN Red List of Threatened Species™ is produced and managed by the IUCN Global Species Programme, the IUCN Species Survival Commission (SSC) and The IUCN Red List Partnership. The IUCN Red List Partners are: BirdLife International; Botanic Gardens Conservation International; Conservation International; Microsoft; NatureServe; Royal Botanic Gardens, Kew; Sapienza University of Rome; Texas A&M University; Wildscreen; and Zoological Society of London. If you see any errors or have any questions or suggestions on what is shown in this document, please provide us with feedback so that we can correct or extend the information -

The Florida Panther: a Story of Conflict, Connections and Coexistence
The Florida Panther: A Story of Conflict, Connections and Coexistence Laurie Macdonald Florida Director DefendersDefenders of of Wildlife Wildlife Endangered – US Endangered Species Act of 1973 Population estimate in 1970s = 12-20 Genetic reinvigoration program 1995 Population estimate today = 100-180 Defenders of Wildlife Florida’s panther story Figure prepared by the Florida Fish & Wildlife Conservation Commission Once upon a time…. Sustainable coexistence? Little or no management Abundant Over- Conflict exploited management Photo Credit: Cory, Charles B. Hunting and Fishing in Florida. New York: Arno Press 1970 Rare More numerous Recovery Rebounding management Florida now and future… • 19 million people • 34.7 million acres 3 Foot Sea Level Rise Defenders of Wildlife Chances of successful recovery will be the greatest if the panther is able to travel north on its own and resettle its historic home. Defenders of Wildlife * Core habitat * Connectivity * Coexistence Defenders of Wildlife Conservation of Core and Corridor Habitat Habitat Protection at the Landscape Level Defenders of Wildlife Defenders of Wildlife Amendment 1 – Florida’s Water and Land Legacy State Constitutional Amendment Vote November 4th, 2014 Greater Everglades Refuge Planning Areas Connectivity: Corridors, Linkages, Networks Defenders of Wildlife Defenders of Wildlife ©Mark Lotz, Florida Fish and Wildlife Conservation Commission Defenders of Wildlife Florida Black Bear (Ursus americanus floridanus) American Alligator (Alligator mississippiensis) Defenders of Wildlife -

First Record of Hose's Civet Diplogale Hosei from Indonesia
First record of Hose’s Civet Diplogale hosei from Indonesia, and records of other carnivores in the Schwaner Mountains, Central Kalimantan, Indonesia Hiromitsu SAMEJIMA1 and Gono SEMIADI2 Abstract One of the least-recorded carnivores in Borneo, Hose’s Civet Diplogale hosei , was filmed twice in a logging concession, the Katingan–Seruyan Block of Sari Bumi Kusuma Corporation, in the Schwaner Mountains, upper Seruyan River catchment, Central Kalimantan. This, the first record of this species in Indonesia, is about 500 km southwest of its previously known distribution (northern Borneo: Sarawak, Sabah and Brunei). Filmed at 325The m a.s.l., IUCN these Red List records of Threatened are below Species the previously known altitudinal range (450–1,800Prionailurus m). This preliminary planiceps survey forPardofelis medium badia and large and Otter mammals, Civet Cynogalerunning 100bennettii camera-traps in 10 plots for one (Bandedyear, identified Civet Hemigalus in this concession derbyanus 17 carnivores, Arctictis including, binturong on Neofelis diardi, three Endangered Pardofe species- lis(Flat-headed marmorata Cat and Sun Bear Helarctos malayanus, Bay Cat . ) and six Vulnerable species , Binturong , Sunda Clouded Leopard , Marbled Cat Keywords Cynogale bennettii, as well, Pardofelis as Hose’s badia Civet), Prionailurus planiceps Catatan: PertamaBorneo, camera-trapping, mengenai Musang Gunung Diplogale hosei di Indonesia, serta, sustainable karnivora forest management lainnya di daerah Pegunungan Schwaner, Kalimantan Tengah Abstrak Diplogale hosei Salah satu jenis karnivora yang jarang dijumpai di Borneo, Musang Gunung, , telah terekam dua kali di daerah- konsesi hutan Blok Katingan–Seruyan- PT. Sari Bumi Kusuma, Pegunungan Schwaner, di sekitar hulu Sungai Seruya, Kalimantan Tengah. Ini merupakan catatan pertama spesies tersebut terdapat di Indonesia, sekitar 500 km dari batas sebaran yang diketa hui saat ini (Sarawak, Sabah, Brunei). -

1 the Origin and Evolution of the Domestic Cat
1 The Origin and Evolution of the Domestic Cat There are approximately 40 different species of the cat family, classification Felidae (Table 1.1), all of which are descended from a leopard-like predator Pseudaelurus that existed in South-east Asia around 11 million years ago (O’Brien and Johnson, 2007). Other than the domestic cat, the most well known of the Felidae are the big cats such as lions, tigers and panthers, sub-classification Panthera. But the cat family also includes a large number of small cats, including a group commonly known as the wildcats, sub-classification Felis silvestris (Table 1.2). Physical similarity suggests that the domestic cat (Felis silvestris catus) originally derived from one or more than one of these small wildcats. DNA examination shows that it is most closely related to the African wildcat (Felis silvestris lybica), which has almost identical DNA, indicating that the African wildcat is the domestic cat’s primary ancestor (Lipinski et al., 2008). The African Wildcat The African wildcat is still in existence today and is a solitary and highly territorial animal indigenous to areas of North Africa and the Near East, the region where domestication of the cat is believed to have first taken place (Driscoll et al., 2007; Faure and Kitchener, 2009). It is primarily a nocturnal hunter that preys mainly on rodents but it will also eat insects, reptiles and other mammals including the young of small antelopes. Also known as the Arabian or North African wildcat, it is similar in appearance to a domestic tabby, with a striped grey/sandy-coloured coat, but is slightly larger and with longer legs (Fig. -

Bay Cat 1 Bay Cat
Bay Cat 1 Bay Cat Bay Cat Conservation status [1] Endangered (IUCN 3.1) Scientific classification Kingdom: Animalia Phylum: Chordata Class: Mammalia Order: Carnivora Family: Felidae Genus: Pardofelis Species: P. badia Binomial name Pardofelis badia[1] (Gray, 1874) Bay Cat 2 [2] Blue dots indicate bay cat records from 2003 to 2005. The Bay Cat (Pardofelis badia syn. Catopuma badia), also known as Bornean Cat, Bornean Bay Cat, Bornean Marbled Cat, is a wild cat endemic to the island of Borneo that appears relatively rare compared to sympatric felids, based on the paucity of historical as well as recent records. In 2002, the IUCN classified the forest-dependent species as endangered because of a projected population decline by more than 20% by 2020 due to habitat loss. As of 2007, the effective population size was suspected to be below 2,500 mature individuals.[1] Bay cats have historically been recorded as rare and today seem to occur at relatively low density, even in pristine habitat.[3] Characteristics The bay cat is much larger than the Asian golden cat. Its fur is of a bright chestnut-colour, rather paler beneath, the limbs and the tail being rather paler and redder. The tail is elongate, tapering at the end, with a white central streak occupying the hinder half of the lower side, gradually becoming wider and of a purer white towards the tip, which has a small black spot at its upper end. The ears are rounded, covered with a short blackish-brown fur at the outer side, paler brown within and with a narrow brown margin.[4] [4] Illustration of a bay cat In the years between 1874 to 2004, only 12 specimens were measured. -

Pallas's Cat Status Review & Conservation Strategy
ISSN 1027-2992 I Special Issue I N° 13 | Spring 2019 Pallas'sCAT cat Status Reviewnews & Conservation Strategy 02 CATnews is the newsletter of the Cat Specialist Group, Editors: Christine & Urs Breitenmoser a component of the Species Survival Commission SSC of the Co-chairs IUCN/SSC International Union for Conservation of Nature (IUCN). It is pub- Cat Specialist Group lished twice a year, and is available to members and the Friends of KORA, Thunstrasse 31, 3074 Muri, the Cat Group. Switzerland Tel ++41(31) 951 90 20 For joining the Friends of the Cat Group please contact Fax ++41(31) 951 90 40 Christine Breitenmoser at [email protected] <[email protected]> <[email protected]> Original contributions and short notes about wild cats are welcome Send contributions and observations to Associate Editors: Tabea Lanz [email protected]. Guidelines for authors are available at www.catsg.org/catnews This Special Issue of CATnews has been produced with Cover Photo: Camera trap picture of manul in the support from the Taiwan Council of Agriculture's Forestry Bureau, Kotbas Hills, Kazakhstan, 20. July 2016 Fondation Segré, AZA Felid TAG and Zoo Leipzig. (Photo A. Barashkova, I Smelansky, Sibecocenter) Design: barbara surber, werk’sdesign gmbh Layout: Tabea Lanz and Christine Breitenmoser Print: Stämpfli AG, Bern, Switzerland ISSN 1027-2992 © IUCN SSC Cat Specialist Group The designation of the geographical entities in this publication, and the representation of the material, do not imply the expression of any opinion whatsoever on the part of the IUCN concerning the legal status of any country, territory, or area, or its authorities, or concerning the delimitation of its frontiers or boundaries. -

Savannah Cat’ ‘Savannah the Including Serval Hybrids Felis Catus (Domestic Cat), (Serval) and (Serval) Hybrids Of
Invasive animal risk assessment Biosecurity Queensland Agriculture Fisheries and Department of Serval hybrids Hybrids of Leptailurus serval (serval) and Felis catus (domestic cat), including the ‘savannah cat’ Anna Markula, Martin Hannan-Jones and Steve Csurhes First published 2009 Updated 2016 © State of Queensland, 2016. The Queensland Government supports and encourages the dissemination and exchange of its information. The copyright in this publication is licensed under a Creative Commons Attribution 3.0 Australia (CC BY) licence. You must keep intact the copyright notice and attribute the State of Queensland as the source of the publication. Note: Some content in this publication may have different licence terms as indicated. For more information on this licence visit http://creativecommons.org/licenses/ by/3.0/au/deed.en" http://creativecommons.org/licenses/by/3.0/au/deed.en Front cover: Close-up of a 4-month old F1 Savannah cat. Note the occelli on the back of the relaxed ears, and the tear-stain markings which run down the side of the nose. Photo: Jason Douglas. Image from Wikimedia Commons under a Public Domain Licence. Invasive animal risk assessment: Savannah cat Felis catus (hybrid of Leptailurus serval) 2 Contents Introduction 4 Identity of taxa under review 5 Identification of hybrids 8 Description 10 Biology 11 Life history 11 Savannah cat breed history 11 Behaviour 12 Diet 12 Predators and diseases 12 Legal status of serval hybrids including savannah cats (overseas) 13 Legal status of serval hybrids including savannah cats -

Origin of the Egyptian Domestic Cat
UPTEC X 12 012 Examensarbete 30 hp Juni 2012 Origin of the Egyptian Domestic Cat Carolin Johansson Molecular Biotechnology Programme Uppsala University School of Engineering UPTEC X 12 012 Date of issue 2012-06 Author Carolin Johansson Title (English) Origin of the Egyptian Domestic Cat Title (Swedish) Abstract This study presents mitochondrial genome sequences from 22 Egyptian house cats with the aim of resolving the uncertain origin of the contemporary world-wide population of Domestic cats. Together with data from earlier studies it has been possible to confirm some of the previously suggested haplotype identifications and phylogeny of the Domestic cat lineage. Moreover, by applying a molecular clock, it is proposed that the Domestic cat lineage has experienced several expansions representing domestication and/or breeding in pre-historical and historical times, seemingly in concordance with theories of a domestication origin in the Neolithic Middle East and in Pharaonic Egypt. In addition, the present study also demonstrates the possibility of retrieving long polynucleotide sequences from hair shafts and a time-efficient way to amplify a complete feline mitochondrial genome. Keywords Feline domestication, cat in ancient Egypt, mitochondrial genome, Felis silvestris libyca Supervisors Anders Götherström Uppsala University Scientific reviewer Jan Storå Stockholm University Project name Sponsors Language Security English Classification ISSN 1401-2138 Supplementary bibliographical information Pages 123 Biology Education Centre Biomedical Center Husargatan 3 Uppsala Box 592 S-75124 Uppsala Tel +46 (0)18 4710000 Fax +46 (0)18 471 4687 Origin of the Egyptian Domestic Cat Carolin Johansson Populärvetenskaplig sammanfattning Det är inte sedan tidigare känt exakt hur, när och var tamkatten domesticerades. -

Ecologists Warn of Japanese Badger Cull 'Crisis' : Nature News & Comment
NATURE | NEWS Ecologists warn of Japanese badger cull 'crisis' Population crash feared amid a fad for badger meat. Tim Hornyak 09 June 2017 alpsdake/CC BY-SA 4.0 The Japanese badger (Meles anakuma). On Japan’s Kyushu Island, farmers regularly trap and spear local badgers, which are regarded as pests. But ecologists say the practice is getting out of hand. In Kyushu’s Kagoshima Prefecture, they note, killings spiked from a few hundred to more than 4,000 last year — and that might lead to a population crash. “If the cull continues at this pace, there’s a possibility the Japanese badger could become extinct,” says Yayoi Kaneko, an ecologist at Tokyo University of Agriculture and Technology. A culinary fad for badger meat in Japan's restaurants is also taking off, although it's unclear if that is driving the culls, or is a response to the ready supply. Japan’s government should intervene in the cull and take scientific advice on whether it is sustainable, the scientists say. Ecological crisis Kaneko and two other ecologists, Christina Buesching and Chris Newman at the University of Oxford, UK, first raised their concerns in a correspondence published in Nature on 13 April 1. They warned that the rise in killings could lead to an “ecological crisis” unfolding, and say that the cull is being carried out “without scientific advice or strategic planning”. The Japanese badger (Meles Related stories Related stories anakuma) is endemic to Japan. It is • Japan: Unjustified killing • Japan: Unjustified killing smaller than its European of badgers in Kyushu of badgers in Kyushu counterpart and has less-distinct facial stripes.