Extended and Continuous Decline in Effective Population Size Results in Low Genomic Diversity in the World’S Rarest Hyena Species, the Brown Hyena Michael V
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
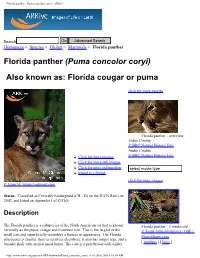
Florida Panther - Puma Concolor Coryi - Arkive
Florida panther - Puma concolor coryi - ARKive Search Homepage > Species > Global > Mammals > Florida panther Florida panther (Puma concolor coryi) Also known as: Florida cougar or puma click for more movies Florida panther - overview Video Credits: © BBC Natural History Unit Audio Credits: © BBC Natural History Unit ● Click for more movies ● Click for more still images ● Click for more information ● Email to a friend click for more images © Lynn M. Stone / naturepl.com Status: Classified as Critically Endangered (CR - D) on the IUCN Red List 2002, and listed on Appendix I of CITES. Description The Florida panther is a subspecies of the North American cat that is known Florida panther - 3 weeks old variously as the puma, cougar and mountain lion. This is the largest of the © Frank Schneidermeyer / OSF / small cats and superficially resembles a lioness in appearance. The Florida Photolibrary.com subspecies is smaller than its relatives elsewhere; it also has longer legs, and a [ medium ] [ large ] broader skull with arched nasal bones. The coat is a pale brown with whiter http://www.arkive.org/species/GES/mammals/Puma_concolor_coryi/ (1 of 2)4/6/2005 8:16:04 AM Florida panther - Puma concolor coryi - ARKive underparts and a black tip at the end of the long tail. Infants have a spotted coat and blue eyes. Florida panthers often have crooked ends to their tails, and whorls of hair on their backs; these are thought not to be characteristic of the subspecies however, and may be signs of inbreeding. Click for more information Florida panther - 5 months old © Bob Bennett / OSF / Photolibrary.com [ medium ] [ large ] © Wildscreen 2004 By using this website you agree to the Terms of Use About ARKive | Competition | Contact | Newsletter | FAQ | Links http://www.arkive.org/species/GES/mammals/Puma_concolor_coryi/ (2 of 2)4/6/2005 8:16:04 AM. -

The Florida Panther: a Story of Conflict, Connections and Coexistence
The Florida Panther: A Story of Conflict, Connections and Coexistence Laurie Macdonald Florida Director DefendersDefenders of of Wildlife Wildlife Endangered – US Endangered Species Act of 1973 Population estimate in 1970s = 12-20 Genetic reinvigoration program 1995 Population estimate today = 100-180 Defenders of Wildlife Florida’s panther story Figure prepared by the Florida Fish & Wildlife Conservation Commission Once upon a time…. Sustainable coexistence? Little or no management Abundant Over- Conflict exploited management Photo Credit: Cory, Charles B. Hunting and Fishing in Florida. New York: Arno Press 1970 Rare More numerous Recovery Rebounding management Florida now and future… • 19 million people • 34.7 million acres 3 Foot Sea Level Rise Defenders of Wildlife Chances of successful recovery will be the greatest if the panther is able to travel north on its own and resettle its historic home. Defenders of Wildlife * Core habitat * Connectivity * Coexistence Defenders of Wildlife Conservation of Core and Corridor Habitat Habitat Protection at the Landscape Level Defenders of Wildlife Defenders of Wildlife Amendment 1 – Florida’s Water and Land Legacy State Constitutional Amendment Vote November 4th, 2014 Greater Everglades Refuge Planning Areas Connectivity: Corridors, Linkages, Networks Defenders of Wildlife Defenders of Wildlife ©Mark Lotz, Florida Fish and Wildlife Conservation Commission Defenders of Wildlife Florida Black Bear (Ursus americanus floridanus) American Alligator (Alligator mississippiensis) Defenders of Wildlife -

Florida Panthers Typically Will a Confrontation.Avoid Give Them Escape
You live in Florida panther country If you see a Florida panther A guide to living with Florida panthers are reclusive and rarely seen by people. The Florida panther moves primarily at night. The They normally live in remote, undeveloped areas. chances of seeing a panther are slim. But if you live in printed on recycled paper MyFWC.com/Panther Tallahassee,32399-1600 FL 620 S. MeridianStreet Conservation Commission Florida Fish and Wildlife FishFlorida Wildlife and However, as the number of people in southern Florida Florida panther country, you need to know what to do if grows, there is an increased chance of an encounter you see one. Florida with a Florida panther. Keep children within sight and close to you. Pick This brochure contains some guidelines to help you live up any small children so they don’t panic and run. Panthers safely in Florida panther country. Try to do this without bending over or turning away from the Florida panther. Give them space. Florida panthers typically will avoid a confrontation. Give them a way to escape. Do not run. Running may stimulate a panther’s instinct to chase. Stand and face the animal. Make eye contact to let the panther know you are aware of its presence. Avoid crouching or bending over. Squatting or bending makes you look smaller, resembling a prey- sized animal. Appear larger. Make gestures that indicate you are not prey and that you may be a danger to the panther. Raise your arms. Open your jacket. Throw stones, branches or whatever you can reach without crouching or turning your back. -

Revealed Via Genomic Assessment of Felid Cansines
Evolutionary and Functional Impacts of Short Interspersed Nuclear Elements (SINEs) Revealed via Genomic Assessment of Felid CanSINEs By Kathryn B. Walters-Conte B. S., May 2000, University of Maryland, College Park M. S., May 2002, The George Washington University A Dissertation Submitted to The Faculty of Columbian College of Arts and Sciences of The George Washington University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy May 15 th , 2011 Dissertation Directed By Diana L.E. Johnson Associate Professor of Biology Jill Pecon-Slattery Staff Scientist, National Cancer Institute . The Columbian College of Arts and Sciences of The George Washington University certifies that Kathryn Walters-Conte has passed the Final Examination for the degree of Doctor of Philosophy as of March 24 th , 2011. This is the final and approved form of the dissertation. Evolutionary and Functional Impacts of Short Interspersed Nuclear Elements (SINEs) Revealed via Genomic Assessment of Felid CanSINEs Kathryn Walters-Conte Dissertation Research Committee: Diana L.E. Johnson, Associate Professor of Biology, Dissertation Co-Director Jill Pecon-Slattery, Staff Scientist, National Cancer Institute, Dissertation Co-Director Diana Lipscomb, Ronald Weintraub Chair and Professor, Committee Member Marc W. Allard, Research Microbiologist, U.S. Food and Drug Administration, Committee Member ii Acknowledgements I would like to first thank my advisor and collaborator, Dr. Jill Pecon-Slattery, at the National Cancer Institute of the National Institutes of Health, for generously permitting me to join her research group. Without her mentorship this dissertation would never have been possible. I would also like to express gratitude to my advisor at the George Washington University, Dr. -

The Role of Big Cypress National Preserve (Big Cypress) in Florida Panther Recovery Has Evolved As Research Has Replaced Specula
FLORIDA PANTHER (Puma concolor coryi) RESEARCH AND MONITORING IN BIG CYPRESS NATIONAL PRESERVE 2009-2010 ANNUAL REPORT Photo of FP175 by Ralph Arwood Submitted to U. S. Fish and Wildlife Service (Endangered Species Permit TE146761-1 and Florida Fish and Wildlife Conservation Commission (Special Purpose Permit WX08654) December 31, 2010 Prepared by National Park Service staff Deborah Jansen, Project Leader John Kellam, Biological Technician Annette Johnson, Biological Technician Table of Contents Abstract .................................................................................................................................................... 1 Report Background ................................................................................................................................... 2 Statement of Purpose ................................................................................................................................ 2 Project Goals ............................................................................................................................................ 3 Study Area ............................................................................................................................................... 4 Methods ................................................................................................................................................... 5 Study Area Sampling ........................................................................................................................ -

Florida Panther Puma Concolor Coryi
Florida Panther Puma concolor coryi he Florida panther, a subspecies of mountain lion, is Federal Status: Endangered (March 11, 1967) one of the most endangered large mammals in the Critical Habitat: None Designated Tworld. It is also Floridas state animal. A small Florida Status: Endangered population in South Florida,estimated to number between 30 and 50 adults (30 to 80 total individuals), represents the only Recovery Plan Status: Contribution (May 1999) known remaining wild population of an animal that once Geographic Coverage: South Florida ranged throughout most of the southeastern United States from Arkansas and Louisiana eastward across Mississippi, Alabama, Georgia, Florida and parts of South Carolina and Figure 1. County distribution of the Florida panther since 1981, based on radiotelemetry data. Tennessee. The panther presently occupies one of the least developed areas in the eastern United States; a contiguous system of large private ranches and public conservation lands in Broward, Collier, Glades, Hendry, Lee, Miami-Dade, Monroe, and Palm Beach counties totaling more than 809,400 ha. Geographic isolation, habitat loss, population decline, and associated inbreeding have resulted in a significant loss of genetic variability and overall health of the Florida panther population. Natural gene exchange ceased when the panther became geographically isolated from other subspecies of Puma concolor about a century ago. Population viability projections have concluded that, under current demographic and genetic conditions, the panther would probably become extinct within two to four decades. A genetic management program was implemented with the release of eight female Texas cougars (Puma concolor stanleyana) into South Florida in 1995 (refer to the Management section for a discussion of this program). -
Florida Panther
U.S. Fish & Wildlife Service Florida Panther National Wildlife FloridaFlorida PantherPanther NationalNational WildlifeWildlife RefugeRefuge isis oneone ofof overover 540540 NationalNational WildlifeWildlife RefugesRefuges administeredadministered byby thethe U.S.U.S. FishFish andand WildlifeWildlife Service.Service. TheThe missionmission ofof thethe NationalNational WildlifeWildlife RefugeRefuge SystemSystem isis toto administeradminister aa nationalnational networknetwork ofof landslands forfor thethe conservation,conservation, management,management, andand wherewhere appropriate,appropriate, thethe restorationrestoration ofof fish,fish, wildlife,wildlife, andand plantplant resourcesresources andand theirtheir habitatshabitats withinwithin thethe UnitedUnited StatesStates forfor thethe benefitbenefit ofof presentpresent andand futurefuture generationsgenerations ofof Americans.Americans. The National Wildlife Refuge System FloridaFlorida PantherPanther For over a century, the National Wildlife Refuge System has been the hidden jewel among our nation’s public NationalNational WildlifeWildlife lands. The Refuge System, since 1903 This blue goose, has grown to nearly 100 million acres designed by Ding and more than 540 refuges. RefugeRefuge isis oneone ofof overover Darling, has become a symbol 540540 NationalNational WildlifeWildlife of the Refuge Florida Panther National Wildlife System. Refuge Was established in June 1989 to RefugesRefuges administeredadministered protect the Florida panther and its habitat. The 26,400 acres are located byby -

Kautz Et Al., How Much Is Enough? Landscape-Scale Conservation for the Florida Panther
BIOLOGICAL CONSERVATION 130 (2006) 118– 133 available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/biocon How much is enough? Landscape-scale conservation for the Florida panther Randy Kautza,*, Robert Kawulaa, Thomas Hoctorb, Jane Comiskeyc, Deborah Jansend, Dawn Jenningse, John Kasbohmf, Frank Mazzottig, Roy McBrideh, Larry Richardsoni, Karen Rootj aFlorida Fish and Wildlife Conservation Commission, 620 South Meridian Street, Tallahassee, FL 32399-1600, USA bUniversity of Florida, Department of Landscape Architecture, P.O. Box 115704, Gainesville, FL 32611-5704, USA cThe Institute for Environmental Modeling, Department of Ecology and Evolutionary Biology, University of Tennessee, Knoxville, TN 37996, USA dBig Cypress National Preserve, 33100 Tamiami Trail East, Ochopee, FL 34141, USA eUS Fish and Wildlife Service, 6620 South Point Drive South, Suite 310, Jacksonville, FL 32216, USA fUS Fish and Wildlife Service, 1502 S.E. Kings Bay Drive, Crystal River, FL 34429, USA gFort Lauderdale Research and Education Center, Department of Wildlife Ecology and Conservation, 3205 College Avenue, Davie, FL 33314, USA hBox 178, Ochopee, FL 34141, USA iUS Fish and Wildlife Service, Florida Panther National Wildlife Refuge, 3860 Tollgate Boulevard, Suite 300, Naples, FL 34114, USA jDepartment of Biology, Bowling Green State University, Bowling Green, OH 43403-0212, USA ARTICLE INFO ABSTRACT Article history: The Florida panther (Puma concolor coryi) is an endangered, wide-ranging predator whose Received 10 February 2005 habitat needs conflict with a rapidly growing human population. Our goal was to identify Received in revised form specific regions of the south Florida landscape that are of high conservation value to sup- 29 November 2005 port a self-sustaining panther population. -

Long Extinct Eastern Cougar to Be Removed from Endangered Species List Correcting Lingering Anomaly
News bulletin Media contact: 01/22/2018 Meagan Racey, 413-253-8558, [email protected] (For the Southeast) Phil Kloer, 404-679-7299, [email protected] Long Extinct Eastern Cougar to be Removed from Endangered Species List Correcting Lingering Anomaly Cougars in the East are Florida panthers, released or escaped captives, or animals dispersing from the West The U.S. Fish and Wildlife Service is removing the extinct eastern cougar subspecies (Felis concolor couguar) from the Federal List of Threatened and Endangered Wildlife, correcting a lingering anomaly that listed the species despite it likely having gone extinct many decades before the Endangered Species Act (ESA) was even enacted. Data from researchers, 21 states and Canadian provinces across the subspecies’ former eastern North American range indicate the eastern cougar likely disappeared forever at least 70 years ago. The removal of the extinct subspecies from the endangered species list will take effect February 22, 2018. Extinct animals and plants cannot be protected under the Endangered Species Act, which is meant to recover imperiled wildlife and plants and their habitats. Additionally, under law, the eastern cougar listing cannot be used as a method to protect other cougars. During a 2011 review of the subspecies’ status under the ESA, and subsequent 2015 proposal to delist, no states or provinces provided evidence of the existence of an eastern cougar population, nor did analysis of hundreds of reports from the public suggest otherwise. While many suspected cougar sightings are probably mistakenly identified bobcats or other animals, cougars do occasionally occur in eastern North America, but they are cougars of other subspecies: either Florida panthers, animals dispersing from western populations, or animals that have been released or escaped from captivity. -

Eastern Puma Questions Put Florida Panther in Perilаннаfriday, July 8
7/11/2016 Eastern puma questions put Florida panther in peril Friday, July 8, 2016 www.eenews.net Eastern puma questions put Florida panther in peril Friday, July 8, 2016 www.eenews.net Published: Friday, July 8, 2016 The Eastern puma, which hasn't roamed the Northeast in nearly 80 years, is finally poised to be removed from the endangered species list. The big cat is extinct, right? Not so fast. What if the cougar the inspiration for Penn State's Nittany Lion and countless other mascots didn't exist in the first place? That's what some scientists enlisted by the Fish and Wildlife Service to review the delisting proposal for the Eastern puma are suggesting. They argue that for thousands of years, only one species of cougar roamed North America. All the pumas, mountain lions, panthers and ghost cats that called the Eastern Seaboard home until the early 1900s were all just different names for a nowvanished population of Puma concolor. While these reviewers' comments will be focused on the science underpinning the proposal, Endangered Species Act experts worry their feedback could also have serious implications for the endangered Florida panther another cat that has been long, and perhaps incorrectly, considered a cougar subspecies. "In terms of the scientific literature, there never really was an Eastern cougar," said John Laundré, an adjunct member of the State University of New York, Oswego's biological sciences faculty. "Unfortunately, we're still stuck with these archaic divisions of the cougar genome. We talk about the Florida panther as something that's unique, and it really genetically isn't." http://www.eenews.net/greenwire/stories/1060039970/print 1/3 7/11/2016 Eastern puma questions put Florida panther in peril Friday, July 8, 2016 www.eenews.net Laundré is one of six bigcat researchers who was contacted by the agency in late June to comply with a peer review policy that FWS and the National Marine Fisheries Service, which jointly implement the Endangered Species Act, instituted in 1994. -

Panthers in Peril
CONCEPTS OF COMPREHENSION: VOCABULARY IN CONTEXT 4th GRADE UNIT Reading Passage Panthers in Peril Do Florida panthers have nine lives? Scientists sure hope so! The wild cats are on the brink of extinction, or dying out. During the early 1900s, hundreds of the large, light-brown cats roamed the southeastern United States. By the mid- U.S. Fish and Wildlife Service 2000s, panthers could be found only in The Florida panther is a protected southern Florida. Wildlife officials species. estimated that only between 100 and 160 panthers were left in the Sunshine State1. Squeezed Out What caused the sharp decline in the panther population? Hunting and habitat loss were mainly to blame. During the mid- to late 1900s, millions of people flocked to Florida for its warm climate and inexpensive land. As people built homes and roads, panthers were squeezed out of their habitat. In 1967, Florida panthers were placed on the endangered species list in the United States. The list was created to protect animals that are on the edge of extinction. Road to Recovery Wildlife officials have been working to keep the wild cats out of peril, or danger. Workers have installed fencing along 40 miles of public roads in southwestern Florida to prevent panthers from colliding with cars and other vehicles. 1 Sunshine State: nickname for Florida 1 © 2012 ReadWorks®, Inc. All rights reserved. Article: Copyright © 2007 Weekly Reader Corporation. All rights reserved. Weekly Reader is a registered trademark of Weekly Reader Corporation. Used by permission. CONCEPTS OF COMPREHENSION: VOCABULARY IN CONTEXT 4th GRADE UNIT Reading Passage Officials also have made efforts to save the land where panthers live from further development. -

American Alligator
American alligator Alligator mississippiensis (Photo by FWC) Taxonomic Classification Kingdom: Animalia Phylum: Chordata Class: Reptilia Order: Crocodila Family: Alligatoridae Genus/Species: Alligator mississippiensis Common Name: American alligator Listing Status Federal Status: Threatened (Similarity of Appearance to the American Crocodile) FL Status: Federally-designated Threatened (Similarity of Appearance) FNAI Ranks: G5/S4 (Globally: Demonstrably Secure/State: Apparently Secure). IUCN Status: LC (Least Concern) Physical Description The American alligator is a black reptile that can reach lengths of 13-14.7 feet (4-4.5 meters) and a weight in excess of 1,000 lbs (454 kilograms) (Florida Museum of Natural History, n.d.). A special feature of the alligator is its nostrils. The nostrils are upward facing on the long snout, which allows the alligator to breathe when its body is submerged (Smithsonian National Zoological Park, n.d.). Also, armored plates (scutes) cover the body. Alligators have a vertically flattened tail, and a light yellow throat and belly. Alligators are ectothermic (body temperature is externally regulated) and are generally active when external temperatures are 82-92°F (28-33°C). American Alligator 1 | Page They stop feeding when the external temperature drops to 70°F (21°C), and they will go dormant (inactive) at (55°F) 13°C. While in a dormant stage, they can be found in burrows. Life History Adult alligators are opportunistic feeders (they feed on what is available). Adults feed on small mammals, birds, fish, turtles, and snakes; juveniles feed on small fish, insects, and amphibians. In the Everglades, alligators primarily feed on apple snails (P. Moler pers.