Design of Vaccine Efficacy Trials to Be Used During Public Health Emergencies – Points of Considerations and Key Principles
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Descriptive Statistics (Part 2): Interpreting Study Results
Statistical Notes II Descriptive statistics (Part 2): Interpreting study results A Cook and A Sheikh he previous paper in this series looked at ‘baseline’. Investigations of treatment effects can be descriptive statistics, showing how to use and made in similar fashion by comparisons of disease T interpret fundamental measures such as the probability in treated and untreated patients. mean and standard deviation. Here we continue with descriptive statistics, looking at measures more The relative risk (RR), also sometimes known as specific to medical research. We start by defining the risk ratio, compares the risk of exposed and risk and odds, the two basic measures of disease unexposed subjects, while the odds ratio (OR) probability. Then we show how the effect of a disease compares odds. A relative risk or odds ratio greater risk factor, or a treatment, can be measured using the than one indicates an exposure to be harmful, while relative risk or the odds ratio. Finally we discuss the a value less than one indicates a protective effect. ‘number needed to treat’, a measure derived from the RR = 1.2 means exposed people are 20% more likely relative risk, which has gained popularity because of to be diseased, RR = 1.4 means 40% more likely. its clinical usefulness. Examples from the literature OR = 1.2 means that the odds of disease is 20% higher are used to illustrate important concepts. in exposed people. RISK AND ODDS Among workers at factory two (‘exposed’ workers) The probability of an individual becoming diseased the risk is 13 / 116 = 0.11, compared to an ‘unexposed’ is the risk. -

Exploring Mental Health & COVID-19: How a Pandemic Could Become
Eastern Kentucky University Encompass Honors Theses Student Scholarship Spring 5-2021 Exploring Mental Health & COVID-19: How a Pandemic Could Become America's Next Mental Health Crisis Ashley D. Shofner Eastern Kentucky University, [email protected] Follow this and additional works at: https://encompass.eku.edu/honors_theses Recommended Citation Shofner, Ashley D., "Exploring Mental Health & COVID-19: How a Pandemic Could Become America's Next Mental Health Crisis" (2021). Honors Theses. 834. https://encompass.eku.edu/honors_theses/834 This Open Access Thesis is brought to you for free and open access by the Student Scholarship at Encompass. It has been accepted for inclusion in Honors Theses by an authorized administrator of Encompass. For more information, please contact [email protected]. EASTERN KENTUCKY UNIVERSITY Exploring Mental Health and COVID-19: How a Pandemic Could Become America’s Next Mental Health Crisis Honors Thesis Submitted in Partial Fulfillment of the Requirements of HON 420 Spring 2021 By Ashley D. Shofner Mentor Dr. Molly A. McKinney Associate Professor, Department of Health Promotion and Administration 3 An Abstract Of Exploring Mental Health and COVID-19: How a Pandemic Could Become America’s Next Mental Health Crisis By Ashley D. Shofner Mentor Dr. Molly A. McKinney Associate Professor, Department of Health Promotion and Administration Abstract Description: A pandemic can be described as an epidemic disease that has spread over a large geographical area and has become prevalent in numerous sectors of the globe. In 2020, just over 100 years since our last major pandemic, the 1918 Influenza outbreak, the global community is facing yet another threat: COVID-19. -

Limitations of the Odds Ratio in Gauging the Performance of a Diagnostic, Prognostic, Or Screening Marker
American Journal of Epidemiology Vol. 159, No. 9 Copyright © 2004 by the Johns Hopkins Bloomberg School of Public Health Printed in U.S.A. All rights reserved DOI: 10.1093/aje/kwh101 Limitations of the Odds Ratio in Gauging the Performance of a Diagnostic, Prognostic, or Screening Marker Margaret Sullivan Pepe1,2, Holly Janes2, Gary Longton1, Wendy Leisenring1,2,3, and Polly Newcomb1 1 Division of Public Health Sciences, Fred Hutchinson Cancer Research Center, Seattle, WA. 2 Department of Biostatistics, University of Washington, Seattle, WA. 3 Division of Clinical Research, Fred Hutchinson Cancer Research Center, Seattle, WA. Downloaded from Received for publication June 24, 2003; accepted for publication October 28, 2003. A marker strongly associated with outcome (or disease) is often assumed to be effective for classifying persons http://aje.oxfordjournals.org/ according to their current or future outcome. However, for this assumption to be true, the associated odds ratio must be of a magnitude rarely seen in epidemiologic studies. In this paper, an illustration of the relation between odds ratios and receiver operating characteristic curves shows, for example, that a marker with an odds ratio of as high as 3 is in fact a very poor classification tool. If a marker identifies 10% of controls as positive (false positives) and has an odds ratio of 3, then it will correctly identify only 25% of cases as positive (true positives). The authors illustrate that a single measure of association such as an odds ratio does not meaningfully describe a marker’s ability to classify subjects. Appropriate statistical methods for assessing and reporting the classification power of a marker are described. -

Adaptive Clinical Trials: an Introduction
Adaptive clinical trials: an introduction What are the advantages and disadvantages of adaptive clinical trial designs? How and why were Introduction adaptive clinical Adaptive clinical trial design is trials developed? becoming a hot topic in healthcare research, with some researchers In 2004, the FDA published a report arguing that adaptive trials have the on the problems faced by the potential to get new drugs to market scientific community in developing quicker. In this article, we explain new medical treatments.2 The what adaptive trials are and why they report highlighted that the pace of were developed, and we explore both innovation in biomedical science is the advantages of adaptive designs outstripping the rate of advances and the concerns being raised by in the available technologies and some in the healthcare community. tools for evaluating new treatments. Outdated tools are being used to assess new treatments and there What are adaptive is a critical need to improve the effectiveness and efficiency of clinical trials? clinical trials.2 The current process for developing Adaptive clinical trials enable new treatments is expensive, takes researchers to change an aspect a long time and in some cases, the of a trial design at an interim development process has to be assessment, while controlling stopped after significant amounts the rate of type 1 errors.1 Interim of time and resources have been assessments can help to determine invested.2 In 2006, the FDA published whether a trial design is the most a “Critical Path Opportunities -

Vaccination Certificate Information Version: 21 July 2021
معلومات عن شهادة التطعيم، بالعربية 中文疫苗接种凭证信息 Informations sur le certificat de vaccination en FRANÇAIS Информация о сертификате вакцинации на РУССКОМ ЯЗЫКЕ Información sobre el Certificado de Vacunación en ESPAÑOL UN SYSTEM-WIDE COVID-19 VACCINATION PROGRAMME VACCINATION CERTIFICATE INFORMATION VERSION: 21 JULY 2021 ABOUT THE UN SYSTEM-WIDE COVID-19 VACCINATION PROGRAMME The UN is committed to ensuring the protection of its personnel. Tasked by the Secretary-General, the Department of Operational Support (DOS) is leading a coordinated, UN-system-wide effort to ensure the availability of vaccine to UN personnel, their dependents and implementing partners. The roll-out of the UN System-wide COVID-19 Vaccination Programme (the “Programme”) for UN personnel will provide a significant boost to the ability of personnel to stay and deliver on the Organization's mandates, to support beneficiaries in the communities they serve, and to contribute to our on-going work to recover better together from the pandemic. Information about the Programme is available at: https://www.un.org/en/coronavirus/vaccination ABOUT THE VACCINATION CERTIFICATE Upon administration of the requisite vaccine dosage, a certificate of vaccination is generated through the Programme’s registration platform (the “Platform”). The certificate generated by the Platform uses a standardized format, similar to the one used by the WHO in its “International Certificate of Vaccination or Prophylaxis” (Yellow) vaccination booklet. Note: This certificate may not dispense with travel restrictions put in place by the country(ies) of destination. As the nature of the pandemic continues to rapidly evolve, it is highly advisable to check the latest travel regulations. -

Global Dynamics of a Vaccination Model for Infectious Diseases with Asymptomatic Carriers
MATHEMATICAL BIOSCIENCES doi:10.3934/mbe.2016019 AND ENGINEERING Volume 13, Number 4, August 2016 pp. 813{840 GLOBAL DYNAMICS OF A VACCINATION MODEL FOR INFECTIOUS DISEASES WITH ASYMPTOMATIC CARRIERS Martin Luther Mann Manyombe1;2 and Joseph Mbang1;2 Department of Mathematics, Faculty of Science University of Yaounde 1, P.O. Box 812 Yaounde, Cameroon 1;2;3; Jean Lubuma and Berge Tsanou ∗ Department of Mathematics and Applied Mathematics University of Pretoria, Pretoria 0002, South Africa (Communicated by Abba Gumel) Abstract. In this paper, an epidemic model is investigated for infectious dis- eases that can be transmitted through both the infectious individuals and the asymptomatic carriers (i.e., infected individuals who are contagious but do not show any disease symptoms). We propose a dose-structured vaccination model with multiple transmission pathways. Based on the range of the explic- itly computed basic reproduction number, we prove the global stability of the disease-free when this threshold number is less or equal to the unity. Moreover, whenever it is greater than one, the existence of the unique endemic equilibrium is shown and its global stability is established for the case where the changes of displaying the disease symptoms are independent of the vulnerable classes. Further, the model is shown to exhibit a transcritical bifurcation with the unit basic reproduction number being the bifurcation parameter. The impacts of the asymptomatic carriers and the effectiveness of vaccination on the disease transmission are discussed through through the local and the global sensitivity analyses of the basic reproduction number. Finally, a case study of hepatitis B virus disease (HBV) is considered, with the numerical simulations presented to support the analytical results. -

FDA Oncology Experience in Innovative Adaptive Trial Designs
Innovative Adaptive Trial Designs Rajeshwari Sridhara, Ph.D. Director, Division of Biometrics V Office of Biostatistics, CDER, FDA 9/3/2015 Sridhara - Ovarian cancer workshop 1 Fixed Sample Designs • Patient population, disease assessments, treatment, sample size, hypothesis to be tested, primary outcome measure - all fixed • No change in the design features during the study Adaptive Designs • A study that includes a prospectively planned opportunity for modification of one or more specified aspects of the study design and hypotheses based on analysis of data (interim data) from subjects in the study 9/3/2015 Sridhara - Ovarian cancer workshop 2 Bayesian Designs • In the Bayesian paradigm, the parameter measuring treatment effect is regarded as a random variable • Bayesian inference is based on the posterior distribution (Bayes’ Rule – updated based on observed data) – Outcome adaptive • By definition adaptive design 9/3/2015 Sridhara - Ovarian cancer workshop 3 Adaptive Designs (Frequentist or Bayesian) • Allows for planned design modifications • Modifications based on data accrued in the trial up to the interim time • Unblinded or blinded interim results • Control probability of false positive rate for multiple options • Control operational bias • Assumes independent increments of information 9/3/2015 Sridhara - Ovarian cancer workshop 4 Enrichment Designs – Prognostic or Predictive • Untargeted or All comers design: – post-hoc enrichment, prospective-retrospective designs – Marker evaluation after randomization (example: KRAS in cetuximab -
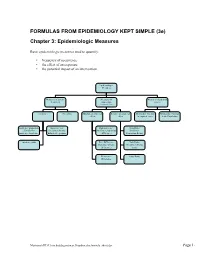
FORMULAS from EPIDEMIOLOGY KEPT SIMPLE (3E) Chapter 3: Epidemiologic Measures
FORMULAS FROM EPIDEMIOLOGY KEPT SIMPLE (3e) Chapter 3: Epidemiologic Measures Basic epidemiologic measures used to quantify: • frequency of occurrence • the effect of an exposure • the potential impact of an intervention. Epidemiologic Measures Measures of disease Measures of Measures of potential frequency association impact (“Measures of Effect”) Incidence Prevalence Absolute measures of Relative measures of Attributable Fraction Attributable Fraction effect effect in exposed cases in the Population Incidence proportion Incidence rate Risk difference Risk Ratio (Cumulative (incidence density, (Incidence proportion (Incidence Incidence, Incidence hazard rate, person- difference) Proportion Ratio) Risk) time rate) Incidence odds Rate Difference Rate Ratio (Incidence density (Incidence density difference) ratio) Prevalence Odds Ratio Difference Macintosh HD:Users:buddygerstman:Dropbox:eks:formula_sheet.doc Page 1 of 7 3.1 Measures of Disease Frequency No. of onsets Incidence Proportion = No. at risk at beginning of follow-up • Also called risk, average risk, and cumulative incidence. • Can be measured in cohorts (closed populations) only. • Requires follow-up of individuals. No. of onsets Incidence Rate = ∑person-time • Also called incidence density and average hazard. • When disease is rare (incidence proportion < 5%), incidence rate ≈ incidence proportion. • In cohorts (closed populations), it is best to sum individual person-time longitudinally. It can also be estimated as Σperson-time ≈ (average population size) × (duration of follow-up). Actuarial adjustments may be needed when the disease outcome is not rare. • In an open populations, Σperson-time ≈ (average population size) × (duration of follow-up). Examples of incidence rates in open populations include: births Crude birth rate (per m) = × m mid-year population size deaths Crude mortality rate (per m) = × m mid-year population size deaths < 1 year of age Infant mortality rate (per m) = × m live births No. -

CEPI and COVID-19 VACCINES
CEPI and COVID-19 VACCINES June 9, 2020 Nicole Lurie, MD, MSPH Strategic Advisor to the CEO and Incident Manager, COVID response team CEPI A world in which epidemics are no longer a threat to humanity CEPI accelerates development of vaccines against emerging infectious diseases and enables equitable access to these vaccines for affected populations during outbreaks 2 Image left slide (right-click to replace image) CEPI Strategic Objectives Preparedness Response Sustainability Advance access to safe and Accelerate the research, Create durable and equitable effective vaccines against development and use of solutions for outbreak emerging infectious diseases vaccines during outbreaks response capacity 3 3 Column slide Small images or graphics can be used to highlight key items. These should always be circular CEPI has multiple investments against its priority pathogens MERS Lassa Nipah Chikungunya Rift Valley fever Disease X 5 vaccine 6 vaccine 4 vaccine 2 vaccine 2 vaccine 3 platform candidates candidates candidates candidates candidates technologies 4 COVID-19 portfolio goals Speed Scale Access Developing Covid-19 vaccines at Scaling up and scaling out vaccine Working with global partners to pandemic speed manufacturing capacity ensure fair allocation of COVID-19 vaccines 5 CEPI vaccine development so far…. 23th May 31st Dec 2019 11th March 12th April 14th Feb First meeting of the ACT WHO notified of pneumonia-like case Wellcome Trust launch First cases reported in WHO declares Accelerator cluster in Wuhan, China COVID-Zero resource Africa -

Statistical Guidance on Reporting Results from Studies Evaluating Diagnostic Tests Document Issued On: March 13, 2007
Guidance for Industry and FDA Staff Statistical Guidance on Reporting Results from Studies Evaluating Diagnostic Tests Document issued on: March 13, 2007 The draft of this document was issued on March 12, 2003. For questions regarding this document, contact Kristen Meier at 240-276-3060, or send an e-mail to [email protected]. U.S. Department of Health and Human Services Food and Drug Administration Center for Devices and Radiological Health Diagnostic Devices Branch Division of Biostatistics Office of Surveillance and Biometrics Contains Nonbinding Recommendations Preface Public Comment Written comments and suggestions may be submitted at any time for Agency consideration to the Division of Dockets Management, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD, 20852. Alternatively, electronic comments may be submitted to http://www.fda.gov/dockets/ecomments. When submitting comments, please refer to Docket No. 2003D-0044. Comments may not be acted upon by the Agency until the document is next revised or updated. Additional Copies Additional copies are available from the Internet at: http://www.fda.gov/cdrh/osb/guidance/1620.pdf. You may also send an e-mail request to [email protected] to receive an electronic copy of the guidance or send a fax request to 240-276-3151 to receive a hard copy. Please use the document number 1620 to identify the guidance you are requesting. Contains Nonbinding Recommendations Table of Contents 1. Background....................................................................................................4 -

Stratification Trees for Adaptive Randomization in Randomized
Stratification Trees for Adaptive Randomization in Randomized Controlled Trials Max Tabord-Meehan Department of Economics Northwestern University [email protected] ⇤ (Click here for most recent version) 26th October 2018 Abstract This paper proposes an adaptive randomization procedure for two-stage randomized con- trolled trials. The method uses data from a first-wave experiment in order to determine how to stratify in a second wave of the experiment, where the objective is to minimize the variance of an estimator for the average treatment e↵ect (ATE). We consider selection from a class of stratified randomization procedures which we call stratification trees: these are procedures whose strata can be represented as decision trees, with di↵ering treatment assignment probabilities across strata. By using the first wave to estimate a stratification tree, we simultaneously select which covariates to use for stratification, how to stratify over these covariates, as well as the assign- ment probabilities within these strata. Our main result shows that using this randomization procedure with an appropriate estimator results in an asymptotic variance which minimizes the variance bound for estimating the ATE, over an optimal stratification of the covariate space. Moreover, by extending techniques developed in Bugni et al. (2018), the results we present are able to accommodate a large class of assignment mechanisms within strata, including stratified block randomization. We also present extensions of the procedure to the setting of multiple treatments, and to the targeting of subgroup-specific e↵ects. In a simulation study, we find that our method is most e↵ective when the response model exhibits some amount of “sparsity” with respect to the covariates, but can be e↵ective in other contexts as well, as long as the first-wave sample size used to estimate the stratification tree is not prohibitively small. -

The 1947 Smallpox Vaccination Campaign in New York City, Revisited
LETTERS The 1947 Smallpox Two days later, epidemiologic contrast, the Army and Navy had investigation indicated that all given almost 800,000 doses, and the Vaccination patients with diagnosed cases were city’s public health laboratories had Campaign in New related and that, in all likelihood, the made the remaining 400,000. York City, Revisited outbreak had been successfully halted During the shortage, the Times through tracing the movements of the noted, “hundreds of eager men, To the Editor: In 1947, millions of various patients and vaccinating any- women, and children queued up at New Yorkers received smallpox vacci- one who had contact with them, so- Bellevue Hospital at dawn, although nations, an accomplishment still appro- called “ring” vaccination (4). Despite vaccinations were not scheduled to priately held up as an example of pub- this halt of the outbreak, the city begin until 10 a.m. At some stations, lic health planning and mobilization. pushed forward. The campaign to “Be the crowds did not take kindly to the Although now mythological, a review sure, be safe, get vaccinated!” had news that the doctors had run out of of the events of April 1947, from proven successful. By city estimate, vaccine and the police had a little dif- copies of The New York Times (1–9), >600,000 persons had received vac- ficulty dispersing a crowd of several tells of a more recognizably human cine in the first week. hundred” outside one vaccine station response: pushing, jawing, deceit, Vaccine side effects, which domi- (5). shortages, surpluses, and perhaps a nate coverage of today’s vaccination On April 17, the situation bright- unusual way of counting vaccinees.