Statistical Guidance on Reporting Results from Studies Evaluating Diagnostic Tests Document Issued On: March 13, 2007
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
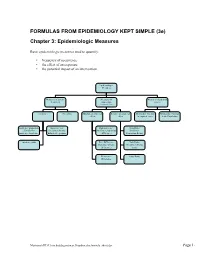
FORMULAS from EPIDEMIOLOGY KEPT SIMPLE (3E) Chapter 3: Epidemiologic Measures
FORMULAS FROM EPIDEMIOLOGY KEPT SIMPLE (3e) Chapter 3: Epidemiologic Measures Basic epidemiologic measures used to quantify: • frequency of occurrence • the effect of an exposure • the potential impact of an intervention. Epidemiologic Measures Measures of disease Measures of Measures of potential frequency association impact (“Measures of Effect”) Incidence Prevalence Absolute measures of Relative measures of Attributable Fraction Attributable Fraction effect effect in exposed cases in the Population Incidence proportion Incidence rate Risk difference Risk Ratio (Cumulative (incidence density, (Incidence proportion (Incidence Incidence, Incidence hazard rate, person- difference) Proportion Ratio) Risk) time rate) Incidence odds Rate Difference Rate Ratio (Incidence density (Incidence density difference) ratio) Prevalence Odds Ratio Difference Macintosh HD:Users:buddygerstman:Dropbox:eks:formula_sheet.doc Page 1 of 7 3.1 Measures of Disease Frequency No. of onsets Incidence Proportion = No. at risk at beginning of follow-up • Also called risk, average risk, and cumulative incidence. • Can be measured in cohorts (closed populations) only. • Requires follow-up of individuals. No. of onsets Incidence Rate = ∑person-time • Also called incidence density and average hazard. • When disease is rare (incidence proportion < 5%), incidence rate ≈ incidence proportion. • In cohorts (closed populations), it is best to sum individual person-time longitudinally. It can also be estimated as Σperson-time ≈ (average population size) × (duration of follow-up). Actuarial adjustments may be needed when the disease outcome is not rare. • In an open populations, Σperson-time ≈ (average population size) × (duration of follow-up). Examples of incidence rates in open populations include: births Crude birth rate (per m) = × m mid-year population size deaths Crude mortality rate (per m) = × m mid-year population size deaths < 1 year of age Infant mortality rate (per m) = × m live births No. -

Estimated HIV Incidence and Prevalence in the United States
Volume 26, Number 1 Estimated HIV Incidence and Prevalence in the United States, 2015–2019 This issue of the HIV Surveillance Supplemental Report is published by the Division of HIV/AIDS Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, Centers for Disease Control and Prevention (CDC), U.S. Department of Health and Human Services, Atlanta, Georgia. Estimates are presented for the incidence and prevalence of HIV infection among adults and adolescents (aged 13 years and older) based on data reported to CDC through December 2020. The HIV Surveillance Supplemental Report is not copyrighted and may be used and reproduced without permission. Citation of the source is, however, appreciated. Suggested citation Centers for Disease Control and Prevention. Estimated HIV incidence and prevalence in the United States, 2015–2019. HIV Surveillance Supplemental Report 2021;26(No. 1). http://www.cdc.gov/ hiv/library/reports/hiv-surveillance.html. Published May 2021. Accessed [date]. On the Web: http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html Confidential information, referrals, and educational material on HIV infection CDC-INFO 1-800-232-4636 (in English, en Español) 1-888-232-6348 (TTY) http://wwwn.cdc.gov/dcs/ContactUs/Form Acknowledgments Publication of this report was made possible by the contributions of the state and territorial health departments and the HIV surveillance programs that provided surveillance data to CDC. This report was prepared by the following staff and contractors of the Division of HIV/AIDS Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, CDC: Laurie Linley, Anna Satcher Johnson, Ruiguang Song, Sherry Hu, Baohua Wu, H. -

Understanding Relative Risk, Odds Ratio, and Related Terms: As Simple As It Can Get Chittaranjan Andrade, MD
Understanding Relative Risk, Odds Ratio, and Related Terms: As Simple as It Can Get Chittaranjan Andrade, MD Each month in his online Introduction column, Dr Andrade Many research papers present findings as odds ratios (ORs) and considers theoretical and relative risks (RRs) as measures of effect size for categorical outcomes. practical ideas in clinical Whereas these and related terms have been well explained in many psychopharmacology articles,1–5 this article presents a version, with examples, that is meant with a view to update the knowledge and skills to be both simple and practical. Readers may note that the explanations of medical practitioners and examples provided apply mostly to randomized controlled trials who treat patients with (RCTs), cohort studies, and case-control studies. Nevertheless, similar psychiatric conditions. principles operate when these concepts are applied in epidemiologic Department of Psychopharmacology, National Institute research. Whereas the terms may be applied slightly differently in of Mental Health and Neurosciences, Bangalore, India different explanatory texts, the general principles are the same. ([email protected]). ABSTRACT Clinical Situation Risk, and related measures of effect size (for Consider a hypothetical RCT in which 76 depressed patients were categorical outcomes) such as relative risks and randomly assigned to receive either venlafaxine (n = 40) or placebo odds ratios, are frequently presented in research (n = 36) for 8 weeks. During the trial, new-onset sexual dysfunction articles. Not all readers know how these statistics was identified in 8 patients treated with venlafaxine and in 3 patients are derived and interpreted, nor are all readers treated with placebo. These results are presented in Table 1. -

Ethics of Vaccine Research
COMMENTARY Ethics of vaccine research Christine Grady Vaccination has attracted controversy at every stage of its development and use. Ethical debates should consider its basic goal, which is to benefit the community at large rather than the individual. accines truly represent one of the mira- include value, validity, fair subject selection, the context in which it will be used and Vcles of modern science. Responsible for favorable risk/benefit ratio, independent acceptable to those who will use it. This reducing morbidity and mortality from sev- review, informed consent and respect for assessment considers details about the pub- eral formidable diseases, vaccines have made enrolled participants. Applying these princi- lic health need (such as the prevalence, bur- substantial contributions to global public ples to vaccine research allows consideration den and natural history of the disease, as health. Generally very safe and effective, vac- of some of the particular challenges inherent well as existing strategies to prevent or con- cines are also an efficient and cost-effective in testing vaccines (Box 1). trol it), the scientific data and possibilities way of preventing disease. Yet, despite their Ethically salient features of clinical vac- (preclinical and clinical data, expected brilliant successes, vaccines have always been cine research include the fact that it involves mechanism of action and immune corre- controversial. Concerns about the safety and healthy subjects, often (or ultimately) chil- lates) and the likely use of the vaccine (who untoward effects of vaccines, about disturb- dren and usually (at least when testing effi- will use and benefit from it, safety, cost, dis- ing the natural order, about compelling indi- cacy) in very large numbers. -

Disease Incidence, Prevalence and Disability
Part 3 Disease incidence, prevalence and disability 9. How many people become sick each year? 28 10. Cancer incidence by site and region 29 11. How many people are sick at any given time? 31 12. Prevalence of moderate and severe disability 31 13. Leading causes of years lost due to disability in 2004 36 World Health Organization 9. How many people become sick each such as diarrhoeal disease or malaria, it is common year? for individuals to be infected repeatedly and have several episodes. For such conditions, the number The “incidence” of a condition is the number of new given in the table is the number of disease episodes, cases in a period of time – usually one year (Table 5). rather than the number of individuals affected. For most conditions in this table, the figure given is It is important to remember that the incidence of the number of individuals who developed the illness a disease or condition measures how many people or problem in 2004. However, for some conditions, are affected by it for the first time over a period of Table 5: Incidence (millions) of selected conditions by WHO region, 2004 Eastern The Mediter- South- Western World Africa Americas ranean Europe East Asia Pacific Tuberculosisa 7.8 1.4 0.4 0.6 0.6 2.8 2.1 HIV infectiona 2.8 1.9 0.2 0.1 0.2 0.2 0.1 Diarrhoeal diseaseb 4 620.4 912.9 543.1 424.9 207.1 1 276.5 1 255.9 Pertussisb 18.4 5.2 1.2 1.6 0.7 7.5 2.1 Measlesa 27.1 5.3 0.0e 1.0 0.2 17.4 3.3 Tetanusa 0.3 0.1 0.0 0.1 0.0 0.1 0.0 Meningitisb 0.7 0.3 0.1 0.1 0.0 0.2 0.1 Malariab 241.3 203.9 2.9 8.6 0.0 23.3 2.7 -

Prevalence and Determinants of Vaccine Hesitancy and Vaccines Recommendation Discrepancies Among General Practitioners in French-Speaking Parts of Belgium
Article Prevalence and Determinants of Vaccine Hesitancy and Vaccines Recommendation Discrepancies among General Practitioners in French-Speaking Parts of Belgium Cathy Gobert 1, Pascal Semaille 2, Thierry Van der Schueren 3 , Pierre Verger 4 and Nicolas Dauby 1,5,6,* 1 Department of Infectious Diseases, CHU Saint-Pierre, Université Libre de Bruxelles (ULB), 1000 Bruxelles, Belgium; [email protected] 2 Department of General Medicine, Université Libre de Bruxelles (ULB), 1070 Bruxelles, Belgium; [email protected] 3 Scientific Society of General Practice, 1060 Bruxelles, Belgium; [email protected] 4 Southeastern Health Regional Observatory (ORS PACA), 13005 Marseille, France; [email protected] 5 School of Public Health, Université Libre de Bruxelles (ULB), 1070 Bruxelles, Belgium 6 Institute for Medical Immunology, Université Libre de Bruxelles (ULB), 1070 Bruxelles, Belgium * Correspondence: [email protected] Abstract: General practitioners (GPs) play a critical role in patient acceptance of vaccination. Vaccine hesitancy (VH) is a growing phenomenon in the general population but also affects GPs. Few data exist on VH among GPs. The objectives of this analysis of a population of GPs in the Belgian Wallonia-Brussels Federation (WBF) were to: (1) determine the prevalence and the features of VH, (2) identify the correlates, and (3) estimate the discrepancy in vaccination’s behaviors between the GPs’ children and the recommendations made to their patients. An online survey was carried out Citation: Gobert, C.; Semaille, P.; Van among the population of general practitioners practicing in the WBF between 7 January and 18 der Schueren, T.; Verger, P.; Dauby, N. March 2020. A hierarchical cluster analysis was carried out based on various dimensions of vaccine Prevalence and Determinants of hesitancy: perception of the risks and the usefulness of vaccines as well as vaccine recommendations Vaccine Hesitancy and Vaccines for their patients. -

EPI Case Study 1 Incidence, Prevalence, and Disease
EPIDEMIOLOGY CASE STUDY 1: Incidence, Prevalence, and Disease Surveillance; Historical Trends in the Epidemiology of M. tuberculosis STUDENT VERSION 1.0 EPI Case Study 1: Incidence, Prevalence, and Disease Surveillance; Historical Trends in the Epidemiology of M. tuberculosis Estimated Time to Complete Exercise: 30 minutes LEARNING OBJECTIVES At the completion of this Case Study, participants should be able to: ¾ Explain why denominators are necessary when comparing changes in morbidity and mortality over time ¾ Distinguish between incidence rates and prevalence ratios ¾ Calculate and interpret cause-specific morbidity and mortality rates ¾ Describe how changes in mortality or morbidity could be due to an artifact rather than a real change ASPH EPIDEMIOLOGY COMPETENCIES ADDRESSED C. 3. Describe a public health problem in terms of magnitude, person, place, and time C. 6. Apply the basic terminology and definitions of epidemiology C. 7. Calculate basic epidemiology measures C. 9. Draw appropriate inference from epidemiologic data C. 10. Evaluate the strengths and limitations of epidemiologic reports ASPH INTERDISCIPLINARY/CROSS-CUTTING COMPETENCIES ADDRESSED F.1. [Communication and Informatics] Describe how the public health information infrastructure is used to collect, process, maintain, and disseminate data J.1. [Professionalism] Discuss sentinel events in the history and development of the public health profession and their relevance for practice in the field L.2. [Systems Thinking] Identify unintended consequences produced by changes made to a public health system This material was developed by the staff at the Global Tuberculosis Institute (GTBI), one of four Regional Training and Medical Consultation Centers funded by the Centers for Disease Control and Prevention. It is published for learning purposes only. -

Likelihood Ratios, Predictive Values, and Post-Test Probabilities The
Likelihood ratios, predictive values, and post-test probabilities The purpose of this quick tutorial is to help you better understand how to use likelihood ratios (LRs), predictive values (PVs), and post-test probabilities in medical writing. The key point is that LRs are calculated differently depending on whether a test has only two possible results (dichotomous), or whether it has more than two possible outcomes (multichotomous). Examples of the latter include a clinical decision rule (CDR) that identifies low, moderate and high-risk groups, or reporting serum ferritin as < 20, 20 – 60, 61 – 100, and > 100 mg/dl. Dichotomous tests A likelihood ratio (LR) for a dichotomous test is defined as the likelihood of a test result in patients with the disease divided by the likelihood of the test result in patients without the disease. For example, the likelihood of abnormal fecal immunochemical test (FIT) in patients with colorectal cancer, divided by the likelihood of an abnormal FIT in those without cancer. A LR close to 1 means that the test result does not change the likelihood of disease or the outcome of interest appreciably. The more the likelihood ratio for a positive test (LR+) is greater than 1, the more likely the disease or outcome. The more a likelihood ratio for a negative test is less than 1, the less likely the disease or outcome. Thus, LRs correspond nicely to the clinical concepts of ruling in and ruling out disease. One suggested way to interpret LR is as follows (Ebell, 2005 Information Mastery AAFP Home Study): > 10 Large -

Epidemiology
5 Defining the current situation – epidemiology Paul R Hunter and Helen Risebro The first step in any economic appraisal or evaluation is to understand the underlying problem being addressed (see Chapter 1). Clearly, such an analysis of drinking-water interventions will have a strong public health element. This chapter discusses the role of epidemiology in identifying the burden of disease1 in a community that may be attributable to lack of access to safe drinking-water or adequate sanitation. In order to determine the scale of the problem, there are three questions to be asked: • What is the burden of disease in the target group? 1 WHO measures the burden of disease in disability-adjusted life years (DALYs). This time-bound measurement combines years of life lost as a result of premature death and years of healthy life lost because of time lived in a status of less than full health. Mortality and morbidity are linked to other indicators such as financial costs. © 2011 World Health Organization (WHO). Valuing Water, Valuing Livelihoods. Edited by John Cameron, Paul Hunter, Paul Jagals and Katherine Pond. Published by IWA Publishing, London, UK. 76 Valuing Water, Valuing Livelihoods • What proportion of the burden of disease is caused by deficiencies in access to drinking-water that are to be remedied by the intervention? • Are there any spin-off livelihood effects that would result from the outcomes of the intervention? This chapter focuses on the first two questions. Specific data challenges related to livelihood analysis are raised in Chapter 6. This chapter aims to assist decision-makers in gathering evidence to enable them to make an informed decision about whether or not there is a public health need for an intervention. -

Prevalence and Intensity of Infection of Soil-Transmitted Helminths in Latin America and the Caribbean Countries Mapping at Second Administrative Level 2000-2010
2011 Prevalence and intensity of infection of Soil-transmitted Helminths in Latin America and the Caribbean Countries Mapping at second administrative level 2000-2010 0 March 18/2011 PAHO NTD Team Pan American Health Organization Communicable Disease Prevention and Control Project “Prevalence and intensity of infection of Soil-transmitted Helminths in Latin America and the Caribbean Countries: Mapping at second administrative level 2000-2010” Washington, D.C.: PAHO © 2011 1. Background 2. Objectives 3. Methodology 4. Outcomes 5. Discussion 6. Conclusions All rights reserved. This document may be reviewed, summarized, cited, reproduced, or translated freely, in part or in its entirety with credit given to the Pan American Health Organization. It cannot be sold or used for commercial purposes. The electronic version of this document can be downloaded from: www.paho.org. The ideas presented in this document are solely the responsibility of the authors. Requests for further information on this publication and other publications produced by Neglected and Parasitic Diseases Group, Communicable Disease Prevention and Control Project, HSD/CD should contact: Parasitic and Neglected Diseases Pan American Health Organization 525 Twenty-third Street, N.W. Washington, DC 20037-2895 www.paho.org. Recommended citation: Saboyá MI, Catalá L, Ault SK, Nicholls RS. Prevalence and intensity of infection of Soil-transmitted Helminths in Latin America and the Caribbean Countries: Mapping at second administrative level 2000-2010. Pan American Health Organization: Washington D.C., 2011. 1 March 18/2011 PAHO NTD Team Acknowledgments The Pan-American Health Organization (PAHO) would like to express special thanks to authors who contributed with copies of their papers directly, when free access was not available. -

Estimating COVID-19 Prevalence and Infection Control Practices Among US Dentists
Original Contributions Estimating COVID-19 prevalence and infection control practices among US dentists Cameron G. Estrich, MPH, PhD; Matthew Mikkelsen, MA; Rachel Morrissey, MA; Maria L. Geisinger, DDS, MS; Effie Ioannidou, DDS, MDS; Marko Vujicic, PhD; Marcelo W.B. Araujo, DDS, MS, PhD ABSTRACT Background. Understanding the risks associated with severe acute respiratory syndrome corona- virus 2 (SARS-CoV-2) transmission during oral health care delivery and assessing mitigation strategies for dental offices are critical to improving patient safety and access to oral health care. Methods. The authors invited licensed US dentists practicing primarily in private practice or public health to participate in a web-based survey in June 2020. Dentists from every US state (n ¼ 2,195) answered questions about COVID-19eassociated symptoms, SARS-CoV-2 infection, mental and physical health conditions, and infection control procedures used in their primary dental practices. Results. Most of the dentists (82.2%) were asymptomatic for 1 month before administration of the survey; 16.6% reported being tested for SARS-CoV-2; and 3.7%, 2.7%, and 0% tested positive via respiratory, blood, and salivary samples, respectively. Among those not tested, 0.3% received a probable COVID-19 diagnosis from a physician. In all, 20 of the 2,195 respondents had been infected with SARS-CoV-2; weighted according to age and location to approximate all US dentists, 0.9% (95% confidence interval, 0.5 to 1.5) had confirmed or probable COVID-19. Dentists reported symptoms of depression (8.6%) and anxiety (19.5%). Enhanced infection control procedures were implemented in 99.7% of dentists’ primary practices, most commonly disinfection, COVID-19 screening, social distancing, and wearing face masks. -

Global Prevalence of Asthma-COPD Overlap (ACO) in the General Population: a Systematic Review and Meta-Analysis
Hosseini et al. Respiratory Research (2019) 20:229 https://doi.org/10.1186/s12931-019-1198-4 REVIEW Open Access Global prevalence of asthma-COPD overlap (ACO) in the general population: a systematic review and meta-analysis Mostafa Hosseini1, Amir Almasi-Hashiani2, Mahdi Sepidarkish3 and Saman Maroufizadeh4* Abstract Background: Asthma-COPD overlap (ACO) is a term that encompasses patients with features of both asthma and COPD. To date, the global prevalence of ACO in the general population remains unknown. The objective of this study was to estimate the prevalence of ACO in the general population using a systematic review and meta- analysis. Methods: A systematic search of ISI Web of Knowledge, MEDLINE/PubMed, and Scopus was performed up to May 2019 to identify studies reporting the prevalence of ACO. Reference lists from identified studies and relevant review articles were also searched. Eligibility criteria were studies reporting the prevalence of ACO, performed in general population, and published in English language. Pooled prevalence of ACO with 95% confidence interval (CI) was calculated using random effects Meta-analysis. Results: A total of 27 studies were included in this meta-analysis. The Cochran Q test and I2 statistics revealed substantial heterogeneity among studies. Based on the random-effects model, the pooled prevalence of ACO was 2.0% (95% CI: 1.4–2.6%) in the general population, 26.5% (95% CI: 19.5–33.6%) among patients with asthma, and 29.6% (95% CI: 19.3–39.9%) among patients with COPD. In addition, for included studies, the global prevalence of asthma-only was 6.2% (95% CI: 5.0–7.4%) and COPD-only was 4.9% (95% CI: 4.3–5.5%).