Noncanonical Nf-Κb Signaling Drives Glioma Invasion By
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Highlights of Glycosylation and Adhesion
Highlights of glycosylation and adhesion related genes involved in myogenesis Vincent Grassot, Anne da Silva, James Saliba, Abderrahman Maftah, Fabrice Dupuy, Jean-Michel Petit To cite this version: Vincent Grassot, Anne da Silva, James Saliba, Abderrahman Maftah, Fabrice Dupuy, et al.. Highlights of glycosylation and adhesion related genes involved in myogenesis. BMC Genomics, BioMed Central, 2014, 15 (1), pp.3-17. 10.1186/1471-2164-15-621. hal-01211986 HAL Id: hal-01211986 https://hal.archives-ouvertes.fr/hal-01211986 Submitted on 27 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Grassot et al. BMC Genomics 2014, 15:621 http://www.biomedcentral.com/1471-2164/15/621 RESEARCH ARTICLE Open Access Highlights of glycosylation and adhesion related genes involved in myogenesis Vincent Grassot, Anne Da Silva, James Saliba, Abderrahman Maftah, Fabrice Dupuy and Jean-Michel Petit* Abstract Background: Myogenesis is initiated by myoblast differentiation and fusion to form myotubes and muscle fibres. A population of myoblasts, known as satellite cells, is responsible for post-natal growth of muscle and for its regeneration. This differentiation requires many changes in cell behaviour and its surrounding environment. -

Cell Adhesion Molecules in Normal Skin and Melanoma
biomolecules Review Cell Adhesion Molecules in Normal Skin and Melanoma Cian D’Arcy and Christina Kiel * Systems Biology Ireland & UCD Charles Institute of Dermatology, School of Medicine, University College Dublin, D04 V1W8 Dublin, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +353-1-716-6344 Abstract: Cell adhesion molecules (CAMs) of the cadherin, integrin, immunoglobulin, and selectin protein families are indispensable for the formation and maintenance of multicellular tissues, espe- cially epithelia. In the epidermis, they are involved in cell–cell contacts and in cellular interactions with the extracellular matrix (ECM), thereby contributing to the structural integrity and barrier for- mation of the skin. Bulk and single cell RNA sequencing data show that >170 CAMs are expressed in the healthy human skin, with high expression levels in melanocytes, keratinocytes, endothelial, and smooth muscle cells. Alterations in expression levels of CAMs are involved in melanoma propagation, interaction with the microenvironment, and metastasis. Recent mechanistic analyses together with protein and gene expression data provide a better picture of the role of CAMs in the context of skin physiology and melanoma. Here, we review progress in the field and discuss molecular mechanisms in light of gene expression profiles, including recent single cell RNA expression information. We highlight key adhesion molecules in melanoma, which can guide the identification of pathways and Citation: D’Arcy, C.; Kiel, C. Cell strategies for novel anti-melanoma therapies. Adhesion Molecules in Normal Skin and Melanoma. Biomolecules 2021, 11, Keywords: cadherins; GTEx consortium; Human Protein Atlas; integrins; melanocytes; single cell 1213. https://doi.org/10.3390/ RNA sequencing; selectins; tumour microenvironment biom11081213 Academic Editor: Sang-Han Lee 1. -

Integrin Alpha 11 in the Regulation of the Myofibroblast Phenotype: Implications for Fibrotic Diseases
Integrin alpha 11 in the regulation of the myofibroblast phenotype: implications for fibrotic diseases The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Bansal, R., S. Nakagawa, S. Yazdani, J. van Baarlen, A. Venkatesh, A. P. Koh, W. Song, et al. 2017. “Integrin alpha 11 in the regulation of the myofibroblast phenotype: implications for fibrotic diseases.” Experimental & Molecular Medicine 49 (11): e396. doi:10.1038/ emm.2017.213. http://dx.doi.org/10.1038/emm.2017.213. Published Version doi:10.1038/emm.2017.213 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:34651853 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA OPEN Experimental & Molecular Medicine (2017) 49, e396; doi:10.1038/emm.2017.213 Official journal of the Korean Society for Biochemistry and Molecular Biology www.nature.com/emm ORIGINAL ARTICLE Integrin alpha 11 in the regulation of the myofibroblast phenotype: implications for fibrotic diseases Ruchi Bansal1, Shigeki Nakagawa2,3, Saleh Yazdani1, Joop van Baarlen4, Anu Venkatesh2,3, Anna P Koh2,3, Won-Min Song2,3, Nicolas Goossens2,3, Hideo Watanabe5, Mary B Beasley3,6, Charles A Powell7, Gert Storm1,8, Naftali Kaminski9, Harry van Goor10, Scott L Friedman2,3, Yujin Hoshida2,3 and Jai Prakash1,11,12 Tissue fibrosis, characterized by excessive accumulation of aberrant extracellular matrix (ECM) produced by myofibroblasts, is a growing cause of mortality worldwide. -

Fibroblasts from the Human Skin Dermo-Hypodermal Junction Are
cells Article Fibroblasts from the Human Skin Dermo-Hypodermal Junction are Distinct from Dermal Papillary and Reticular Fibroblasts and from Mesenchymal Stem Cells and Exhibit a Specific Molecular Profile Related to Extracellular Matrix Organization and Modeling Valérie Haydont 1,*, Véronique Neiveyans 1, Philippe Perez 1, Élodie Busson 2, 2 1, 3,4,5,6, , Jean-Jacques Lataillade , Daniel Asselineau y and Nicolas O. Fortunel y * 1 Advanced Research, L’Oréal Research and Innovation, 93600 Aulnay-sous-Bois, France; [email protected] (V.N.); [email protected] (P.P.); [email protected] (D.A.) 2 Department of Medical and Surgical Assistance to the Armed Forces, French Forces Biomedical Research Institute (IRBA), 91223 CEDEX Brétigny sur Orge, France; [email protected] (É.B.); [email protected] (J.-J.L.) 3 Laboratoire de Génomique et Radiobiologie de la Kératinopoïèse, Institut de Biologie François Jacob, CEA/DRF/IRCM, 91000 Evry, France 4 INSERM U967, 92260 Fontenay-aux-Roses, France 5 Université Paris-Diderot, 75013 Paris 7, France 6 Université Paris-Saclay, 78140 Paris 11, France * Correspondence: [email protected] (V.H.); [email protected] (N.O.F.); Tel.: +33-1-48-68-96-00 (V.H.); +33-1-60-87-34-92 or +33-1-60-87-34-98 (N.O.F.) These authors contributed equally to the work. y Received: 15 December 2019; Accepted: 24 January 2020; Published: 5 February 2020 Abstract: Human skin dermis contains fibroblast subpopulations in which characterization is crucial due to their roles in extracellular matrix (ECM) biology. -

Integrin and Gene Network Analysis Reveals That ITGA5 and ITGB1 Are Prognostic in Non-Small-Cell Lung Cancer
Journal name: OncoTargets and Therapy Article Designation: Original Research Year: 2016 Volume: 9 OncoTargets and Therapy Dovepress Running head verso: Zheng et al Running head recto: ITGA5 and ITGB1 are prognostic in NSCLC open access to scientific and medical research DOI: http://dx.doi.org/10.2147/OTT.S91796 Open Access Full Text Article ORIGINAL RESEARCH Integrin and gene network analysis reveals that ITGA5 and ITGB1 are prognostic in non-small-cell lung cancer Weiqi Zheng Background: Integrin expression has been identified as a prognostic factor in non-small-cell Caihui Jiang lung cancer (NSCLC). This study was aimed at determining the predictive ability of integrins Ruifeng Li and associated genes identified within the molecular network. Patients and methods: A total of 959 patients with NSCLC from The Cancer Genome Atlas Department of Radiation Oncology, Guangqian Hospital, Quanzhou, Fujian, cohorts were enrolled in this study. The expression profile of integrins and related genes were People’s Republic of China obtained from The Cancer Genome Atlas RNAseq database. Clinicopathological characteristics, including age, sex, smoking history, stage, histological subtype, neoadjuvant therapy, radiation therapy, and overall survival (OS), were collected. Cox proportional hazards regression models as well as Kaplan–Meier curves were used to assess the relative factors. Results: In the univariate Cox regression model, ITGA1, ITGA5, ITGA6, ITGB1, ITGB4, and ITGA11 were predictive of NSCLC prognosis. After adjusting for clinical factors, ITGA5 (odds ratio =1.17, 95% confidence interval: 1.05–1.31) andITGB1 (odds ratio =1.31, 95% confidence interval: 1.10–1.55) remained statistically significant. In the gene cluster network analysis, PLAUR, ILK, SPP1, PXN, and CD9, all associated with ITGA5 and ITGB1, were identified as independent predictive factors of OS in NSCLC. -

Integrin Binding Dynamics Modulate Ligand-Specific
bioRxiv preprint doi: https://doi.org/10.1101/570721; this version posted March 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Integrin binding dynamics modulate ligand-specific mechanosensing in mammary gland fibroblasts Martina Lerche1, Alberto Elosegui-Artola2, Camilo Guzmán1, Maria Georgiadou1, Jenny Z. Kechagia2, Donald Gullberg3, Pere Roca-Cusachs2,4, Emilia Peuhu1,5 , and Johanna Ivaska1,6 1Centre for Biotechnology, University of Turku and Åbo Akademi University, 20520 Turku, Finland 2Institute for Bioengineering of Catalonia, Barcelona 08028, Spain 3University of Bergen, Bergen, Norway 4University of Barcelona, Barcelona 08028, Spain 5FICAN West Cancer Research Laboratory, University of Turku and Turku University Hospital, 20520 Turku, Finland 6Department of Biochemistry, University of Turku, 20520 Turku, Finland The link between the modulation of integrin activity and cel- tracellular integrin alpha tails and inhibits integrin activity in lular mechanosensing of tissue rigidity, especially on different vitro and in vivo (5-8). We have previously demonstrated that extracellular matrix ligands, remains poorly understood. Here, stromal SHARPIN deficiency interferes with normal mouse we find that primary mouse mammary gland stromal fibrob- mammary gland development and collagen fiber assembly in lasts (MSFs) are mechanically distinct from previously studied vivo (5). However, how and if SHARPIN mediates integrin- cell types. In particular, MSFs generate high forces at a low dependent mechanotransduction remains unresolved. matrix stiffness, equivalent to the soft mammary gland tissue, supported by maximal focal adhesion maturation, strong actin Collagen is abundant in the mammary gland stroma and stress fiber formation, and myosin phosphorylation. -

Integrin Binding Dynamics Modulate Ligand-Specific Mechanosensing
bioRxiv preprint doi: https://doi.org/10.1101/570721; this version posted December 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Integrin binding dynamics modulate ligand-specific mechanosensing in mammary gland fibroblasts Martina Lerche1, Alberto Elosegui-Artola2, Jenny Z. Kechagia2, Camilo Guzmán1, Maria Georgiadou1, Donald Gullberg3, Pere Roca-Cusachs2, 4, Emilia Peuhu1,5,*, and Johanna Ivaska1, 6, * 1Turku Bioscience Centre, University of Turku and Åbo Akademi University, FI-20520 Turku, Finland 2Institute for Bioengineering of Catalonia, 4University of Barcelona, Barcelona 08028, Spain 3University of Bergen, Bergen, Norway 5Institute of Biomedicine and FICAN West Cancer Research Laboratory, University of Turku, FI-20520 Turku, Finland 6Department of Biochemistry, FI-20520 Turku, Finland *Correspondence: Johanna Ivaska, [email protected] or Emilia Peuhu, [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/570721; this version posted December 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Summary The link between the modulation of integrin activity and cellular mechanosensing of tissue rigidity, especially on different extracellular matrix ligands, remains poorly understood. Here, we find that primary mouse mammary gland stromal fibroblasts (MSFs) are able to spread efficiently on soft collagen-coated substrates, resembling the soft mammary gland tissue. -
Integrins As Therapeutic Targets for Respiratory Diseases
Send Orders for Reprints to [email protected] 714 Current Molecular Medicine 2015, 15, 714-734 Integrins as Therapeutic Targets for Respiratory Diseases C.M. Teoh, S.S.L. Tan and T. Tran* Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore Abstract: Integrins are a large family of transmembrane heterodimeric proteins that constitute the main receptors for extracellular matrix components. Integrins were initially thought to be primarily involved in the maintenance of cell adhesion and tissue integrity. However, it is now appreciated that integrins play important roles in many other biological processes such as cell survival, proliferation, differentiation, migration, cell shape and polarity. Lung cells express numerous combinations and permutations of integrin heterodimers. The complexity and diversity of different integrin heterodimers being implicated in different lung diseases present a major challenge for drug development. Here we provide a comprehensive overview of the current knowledge of integrins from studies in cell culture to integrin knockout mouse models and provide an update of results from clinical trials for which integrins are therapeutic targets with a focus on respiratory diseases (asthma, emphysema, pneumonia, lung cancer, pulmonary fibrosis and sarcoidosis). Keywords: Asthma, emphysema, integrins, lung cancer, lung disease, pulmonary fibrosis, sarcoidosis. INTRODUCTION adhesion kinase (FAK); 3) growth factor receptors, such as epidermal growth factor receptor (EGFR) and Respiratory disease is defined as any disease that platelet-derived growth factor (PDGF); and 4) cytokine could impair lung functions. There are many types of receptors, such as IL-3 receptor to influence cell respiratory diseases which include: 1) asthma and behaviors [6]. -
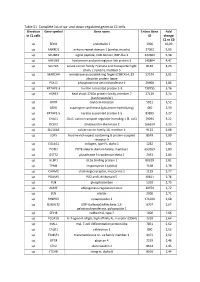
Table S1. Complete List of Up- and Down-Regulated Genes in C1 Cells
Table S1. Complete list of up- and down-regulated genes in C1 cells. Direction Gene symbol Gene name Entrez Gene Fold in C1 cells ID change C1 vs C0 up EDN1 endothelin 1 1906 10,09 up ANKRD1 ankyrin repeat domain 1 (cardiac muscle) 27063 5,95 up SCUBE3 signal peptide, CUB domain, EGF-like 3 222663 5,38 up HAPLN3 hyaluronan and proteoglycan link protein 3 145864 4,47 up SLC7A5 solute carrier family 7 (amino acid transporter light 8140 4,23 chain, L system), member 5 up MARCH4 membrane-associated ring finger (C3HC4) 4, E3 57574 3,91 ubiquitin protein ligase up PSAT1 phosphoserine aminotransferase 1 29968 3,86 up KRTAP2-3 keratin associated protein 2-3 730755 3,76 up HSPB7 heat shock 27kDa protein family, member 7 27129 3,74 (cardiovascular) up OXTR oxytocin receptor 5021 3,52 up ASNS asparagine synthetase (glutamine-hydrolyzing) 440 3,39 up KRTAP1-5 keratin associated protein 1-5 83895 3,27 up CHAC1 ChaC, cation transport regulator homolog 1 (E, coli) 79094 3,22 up DCLK2 doublecortin-like kinase 2 166614 3,15 up SLC16A4 solute carrier family 16, member 4 9122 3,08 up LGR5 leucine-rich repeat containing G protein-coupled 8549 2,99 receptor 5 up COL4A1 collagen, type IV, alpha 1 1282 2,94 up POTEI POTE ankyrin domain family, member I 653269 2,89 up GSTT2 glutathione S-transferase theta 2 2953 2,84 up ULBP1 UL16 binding protein 1 80329 2,81 up TPM1 tropomyosin 1 (alpha) 7168 2,78 up CHRM2 cholinergic receptor, muscarinic 2 1129 2,77 up PDLIM5 PDZ and LIM domain 5 10611 2,76 up PLN phospholamban 5350 2,75 up ADIRF adipogenesis regulatory factor -

Ncomms12502.Pdf
ARTICLE Received 30 Apr 2015 | Accepted 6 Jul 2016 | Published 18 Aug 2016 DOI: 10.1038/ncomms12502 OPEN PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis Katherine Martin1,2,*, James Pritchett1,2,*, Jessica Llewellyn1, Aoibheann F. Mullan1, Varinder S. Athwal1,2, Ross Dobie3, Emma Harvey1, Leo Zeef4, Stuart Farrow1,5, Charles Streuli6, Neil C. Henderson3, Scott L. Friedman7, Neil A. Hanley1,2 & Karen Piper Hanley1,2 Fibrosis due to extracellular matrix (ECM) secretion from myofibroblasts complicates many chronic liver diseases causing scarring and organ failure. Integrin-dependent interaction with scar ECM promotes pro-fibrotic features. However, the pathological intracellular mechanism in liver myofibroblasts is not completely understood, and further insight could enable therapeutic efforts to reverse fibrosis. Here, we show that integrin beta-1, capable of binding integrin alpha-11, regulates the pro-fibrotic phenotype of myofibroblasts. Integrin beta-1 expression is upregulated in pro-fibrotic myofibroblasts in vivo and is required in vitro for production of fibrotic ECM components, myofibroblast proliferation, migration and contrac- tion. Serine/threonine-protein kinase proteins, also known as P21-activated kinase (PAK), and the mechanosensitive factor, Yes-associated protein 1 (YAP-1) are core mediators of pro-fibrotic integrin beta-1 signalling, with YAP-1 capable of perpetuating integrin beta-1 expression. Pharmacological inhibition of either pathway in vivo attenuates liver fibrosis. PAK protein inhibition, in particular, markedly inactivates the pro-fibrotic myofibroblast phenotype, limits scarring from different hepatic insults and represents a new tractable therapeutic target for treating liver fibrosis. 1 Centre for Endocrinology and Diabetes, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, Manchester Academic Health Sciences Centre, Manchester M13 9PT, UK. -

Haplotypes That Include the Integrin Alpha 11 Gene Are Associated with Tick Burden in Cattle BMC Genetics 2010, 11:55
Porto Neto et al. BMC Genetics 2010, 11:55 http://www.biomedcentral.com/1471-2156/11/55 RESEARCH ARTICLE Open Access HaplotypesResearch article that include the integrin alpha 11 gene are associated with tick burden in cattle Laercio R Porto Neto1,2,3, Rowan J Bunch1,2, Blair E Harrison1,2, Kishore C Prayaga1,2 and William Barendse*1,2 Abstract Background: Infestations on cattle by the ectoparasite Boophilus (Rhipicephalus) microplus (cattle tick) impact negatively on animal production systems. Host resistance to tick infestation has a low to moderate heritability in the range 0.13 - 0.64 in Australia. Previous studies identified a QTL on bovine chromosome 10 (BTA10) linked to tick burden in cattle. Results: To confirm these associations, we collected genotypes of 17 SNP from BTA10, including three obtained by sequencing part of the ITGA11 (Integrin alpha 11) gene. Initially, we genotyped 1,055 dairy cattle for the 17 SNP, and then genotyped 557 Brahman and 216 Tropical Composite beef cattle for 11 of the 17 SNP. In total, 7 of the SNP were significantly (P < 0.05) associated with tick burden tested in any of the samples. One SNP, ss161109814, was significantly (P < 0.05) associated with tick burden in both the taurine and the Brahman sample, but the favourable allele was different. Haplotypes for three and for 10 SNP were more significantly (P < 0.001) associated with tick burden than SNP analysed individually. Some of the common haplotypes with the largest sample sizes explained between 1.3% and 1.5% of the residual variance in tick burden.