Classification of Glioma Based on Prognostic Alternative Splicing
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Large-Scale Analysis of Genome and Transcriptome Alterations in Multiple Tumors Unveils Novel Cancer-Relevant Splicing Networks
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Research Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks Endre Sebestyén,1,5 Babita Singh,1,5 Belén Miñana,1,2 Amadís Pagès,1 Francesca Mateo,3 Miguel Angel Pujana,3 Juan Valcárcel,1,2,4 and Eduardo Eyras1,4 1Universitat Pompeu Fabra, E08003 Barcelona, Spain; 2Centre for Genomic Regulation, E08003 Barcelona, Spain; 3Program Against Cancer Therapeutic Resistance (ProCURE), Catalan Institute of Oncology (ICO), Bellvitge Institute for Biomedical Research (IDIBELL), E08908 L’Hospitalet del Llobregat, Spain; 4Catalan Institution for Research and Advanced Studies, E08010 Barcelona, Spain Alternative splicing is regulated by multiple RNA-binding proteins and influences the expression of most eukaryotic genes. However, the role of this process in human disease, and particularly in cancer, is only starting to be unveiled. We system- atically analyzed mutation, copy number, and gene expression patterns of 1348 RNA-binding protein (RBP) genes in 11 solid tumor types, together with alternative splicing changes in these tumors and the enrichment of binding motifs in the alter- natively spliced sequences. Our comprehensive study reveals widespread alterations in the expression of RBP genes, as well as novel mutations and copy number variations in association with multiple alternative splicing changes in cancer drivers and oncogenic pathways. Remarkably, the altered splicing patterns in several tumor types recapitulate those of undifferen- tiated cells. These patterns are predicted to be mainly controlled by MBNL1 and involve multiple cancer drivers, including the mitotic gene NUMA1. We show that NUMA1 alternative splicing induces enhanced cell proliferation and centrosome am- plification in nontumorigenic mammary epithelial cells. -

RBM11 Sirna (H): Sc-91387
SANTA CRUZ BIOTECHNOLOGY, INC. RBM11 siRNA (h): sc-91387 BACKGROUND PRODUCT Proteins containing RNA recognition motifs, including various hnRNP proteins, RBM11 siRNA (h) is a pool of 3 target-specific 19-25 nt siRNAs designed are implicated in the regulation of alternative splicing and protein components to knock down gene expression. Each vial contains 3.3 nmol of lyophilized of snRNPs. The RBM (RNA-binding motif) gene family encodes proteins with siRNA, sufficient for a 10 µM solution once resuspended using protocol an RNA binding motif that have been suggested to play a role in the modu- below. Suitable for 50-100 transfections. Also see RBM11 shRNA Plasmid (h): lation of apoptosis. RBM11 (RNA-binding protein 11) is a 281 amino acid sc-91387-SH and RBM11 shRNA (h) Lentiviral Particles: sc-91387-V as alter- nuclear protein that contains one RNA recognition motif. RBM11 exists as nate gene silencing products. two isoforms produced by alternative splicing and is expressed in testis, kid- For independent verification of RBM11 (h) gene silencing results, we also ney, spleen, brain, spinal cord and mammary gland. The gene that encodes provide the individual siRNA duplex components. Each is available as 3.3 nmol RBM11 maps to chromosome 21, the smallest of the human chromosomes. of lyophilized siRNA. These include: sc-91387A, sc-91387B and sc-91387C. Down syndrome, also known as trisomy 21, is the disease most commonly associated with chromosome 21. Alzheimer’s disease, Jervell and Lange- STORAGE AND RESUSPENSION Nielsen syndrome and amyotrophic lateral sclerosis are also associated with chromosome 21. Store lyophilized siRNA duplex at -20° C with desiccant. -

The Expression of Cold-Inducible RNA-Binding Protein Mrna in Sow Genital Tract Is Modulated by Natural Mating, but Not by Seminal Plasma
International Journal of Molecular Sciences Article The Expression of Cold-Inducible RNA-Binding Protein mRNA in Sow Genital Tract Is Modulated by Natural Mating, but not by Seminal Plasma Jaume Gardela 1,2 , Mateo Ruiz-Conca 1,2 , Cristina A. Martinez 1 , Dominic Wright 3, Manel López-Béjar 2,4, Heriberto Rodriguez-Martinez 1 and Manuel Alvarez-Rodriguez 1,2,* 1 Department of Biomedical and Clinical Sciences (BKV), Division of Children’s and Women Health (BKH), Obstetrics and Gynaecology, Linköping University, 58185 Linköping, Sweden; [email protected] (J.G.); [email protected] (M.R.-C.); [email protected] (C.A.M.); [email protected] (H.R.-M.) 2 Department of Animal Health and Anatomy, Veterinary Faculty, Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain; [email protected] or [email protected] 3 Department of Physics, Chemistry and Biology, Faculty of Science and Engineering, Linköping University, 58185 Linköping, Sweden; [email protected] 4 College of Veterinary Medicine, Western University of Health Sciences, Pomona, CA 91766, USA * Correspondence: [email protected]; Tel.: +46-(0)729427883 Received: 10 June 2020; Accepted: 24 July 2020; Published: 27 July 2020 Abstract: The RNA-binding proteins (RBPs), some of them induced by transient receptor potential (TRP) ion channels, are crucial regulators of RNA function that can contribute to reproductive pathogenesis, including inflammation and immune dysfunction. This study aimed to reveal the influence of spermatozoa, seminal plasma, or natural mating on mRNA expression of RBPs and TRP ion channels in different segments of the internal genital tract of oestrous, preovulatory sows. -

Computational Analyses of Small Molecules Activity from Phenotypic Screens
Computational analyses of small molecules activity from phenotypic screens Azedine Zoufir Hughes Hall This dissertation is submitted for the degree of Doctor of Philosophy July 2018 Declaration This thesis is submitted as the result of my own work and includes nothing which is the outcome of work done in collaboration except where specifically indicated in the text. It is not substantially the same as any that I have submitted, or, is being concurrently submitted for a degree or diploma or other qualification at the University of Cambridge or any other University or similar institution except as declared in the preface and specified in the text. I further state that no substantial part of my dissertation has already been submitted, or, is being concurrently submitted for any such degree, diploma or other qualification at the University of Cambridge or any other University or similar institution except as declared in the Preface and specified in the text. This dissertation does not exceed the word limit of 60,000 words. Azedine Zoufir July 2018 Summary Title: Computational analyses of small molecules activity from phenotypic screens Author: Azedine Zoufir Drug discovery is no longer relying on the one gene-one disease paradigm nor on target-based screening alone to discover new drugs. Phenotypic-based screening is regaining momentum to discover new compounds since those assays provide an environment closer to the physiological state of the disease and allow to better anticipate off-target effects and other factors that can limit the efficacy of the drugs. However, uncovering the mechanism of action of the compounds active in those assays relies on in vitro techniques that are expensive and time- consuming. -

Genomic Evidence of Reactive Oxygen Species Elevation in Papillary Thyroid Carcinoma with Hashimoto Thyroiditis
Endocrine Journal 2015, 62 (10), 857-877 Original Genomic evidence of reactive oxygen species elevation in papillary thyroid carcinoma with Hashimoto thyroiditis Jin Wook Yi1), 2), Ji Yeon Park1), Ji-Youn Sung1), 3), Sang Hyuk Kwak1), 4), Jihan Yu1), 5), Ji Hyun Chang1), 6), Jo-Heon Kim1), 7), Sang Yun Ha1), 8), Eun Kyung Paik1), 9), Woo Seung Lee1), Su-Jin Kim2), Kyu Eun Lee2)* and Ju Han Kim1)* 1) Division of Biomedical Informatics, Seoul National University College of Medicine, Seoul, Korea 2) Department of Surgery, Seoul National University Hospital and College of Medicine, Seoul, Korea 3) Department of Pathology, Kyung Hee University Hospital, Kyung Hee University School of Medicine, Seoul, Korea 4) Kwak Clinic, Okcheon-gun, Chungbuk, Korea 5) Department of Internal Medicine, Uijeongbu St. Mary’s Hospital, Uijeongbu, Korea 6) Department of Radiation Oncology, Seoul St. Mary’s Hospital, Seoul, Korea 7) Department of Pathology, Chonnam National University Hospital, Kwang-Ju, Korea 8) Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea 9) Department of Radiation Oncology, Korea Cancer Center Hospital, Korea Institute of Radiological and Medical Sciences, Seoul, Korea Abstract. Elevated levels of reactive oxygen species (ROS) have been proposed as a risk factor for the development of papillary thyroid carcinoma (PTC) in patients with Hashimoto thyroiditis (HT). However, it has yet to be proven that the total levels of ROS are sufficiently increased to contribute to carcinogenesis. We hypothesized that if the ROS levels were increased in HT, ROS-related genes would also be differently expressed in PTC with HT. To find differentially expressed genes (DEGs) we analyzed data from the Cancer Genomic Atlas, gene expression data from RNA sequencing: 33 from normal thyroid tissue, 232 from PTC without HT, and 60 from PTC with HT. -

Ohnologs in the Human Genome Are Dosage Balanced and Frequently Associated with Disease
Ohnologs in the human genome are dosage balanced and frequently associated with disease Takashi Makino1 and Aoife McLysaght2 Smurfit Institute of Genetics, University of Dublin, Trinity College, Dublin 2, Ireland Edited by Michael Freeling, University of California, Berkeley, CA, and approved April 9, 2010 (received for review December 21, 2009) About 30% of protein-coding genes in the human genome are been duplicated by WGD, subsequent loss of individual genes related through two whole genome duplication (WGD) events. would result in a dosage imbalance due to insufficient gene Although WGD is often credited with great evolutionary impor- product, thus leading to biased retention of dosage-balanced tance, the processes governing the retention of these genes and ohnologs. In fact, evidence for preferential retention of dosage- their biological significance remain unclear. One increasingly pop- balanced genes after WGD is accumulating (4, 7, 11–20). Copy ular hypothesis is that dosage balance constraints are a major number variation [copy number polymorphism (CNV)] describes determinant of duplicate gene retention. We test this hypothesis population level polymorphism of small segmental duplications and show that WGD-duplicated genes (ohnologs) have rarely and is known to directly correlate with gene expression levels (21– experienced subsequent small-scale duplication (SSD) and are also 24). Thus, CNV of dosage-balanced genes is also expected to be refractory to copy number variation (CNV) in human populations deleterious. This model predicts that retained ohnologs should be and are thus likely to be sensitive to relative quantities (i.e., they are enriched for dosage-balanced genes that are resistant to sub- dosage-balanced). -

Morphology, Behavior, and the Sonic Hedgehog Pathway in Mouse Models of Down Syndrome
MORPHOLOGY, BEHAVIOR, AND THE SONIC HEDGEHOG PATHWAY IN MOUSE MODELS OF DOWN SYNDROME by Tara Dutka A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2014 © 2014 Tara Dutka All Rights Reserved Abstract Down Syndrome (DS) is caused by a triplication of human chromosome 21 (Hsa21). Ts65Dn, a mouse model of DS, contains a freely segregating extra chromosome consisting of the distal portion of mouse chromosome 16 (Mmu16), a region orthologous to part of Hsa21, and a non-Hsa21 orthologous region of mouse chromosome 17. All individuals with DS display some level of craniofacial dysmorphology, brain structural and functional changes, and cognitive impairment. Ts65Dn recapitulates these features of DS and aspects of each of these traits have been linked in Ts65Dn to a reduced response to Sonic Hedgehog (SHH) in trisomic cells. Dp(16)1Yey is a new mouse model of DS which has a direct duplication of the entire Hsa21 orthologous region of Mmu16. Dp(16)1Yey’s creators found similar behavioral deficits to those seen in Ts65Dn. We performed a quantitative investigation of the skull and brain of Dp(16)1Yey as compared to Ts65Dn and found that DS-like changes to brain and craniofacial morphology were similar in both models. Our results validate examination of the genetic basis for these phenotypes in Dp(16)1Yey mice and the genetic links for these phenotypes previously found in Ts65Dn , i.e., reduced response to SHH. Further, we hypothesized that if all trisomic cells show a reduced response to SHH, then up-regulation of the SHH pathway might ameliorate multiple phenotypes. -
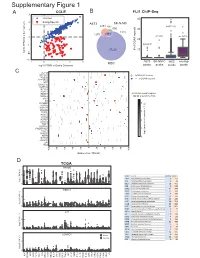
Supplementary Figure 1
6XSSOHPHQWDU\)LJXUH CCLE B FLI1 ChIP-Seq A 3 FRPPRQ 40 (ZLQJ6SHFLILF 2 A673 6.10& 477 Q 690 30 0 UHSHDWV Q Q A 20 í Q 57226 RI**$ ORJ)3.0LQRWKHUFDQFHUV í í A673 6.10& 06& RYHUODS í 0 23 06& ORJ)3.0LQ(ZLQJ6DUFRPD SHDNV SHDNV SHDNV SHDNV & ./) 67($3 (:6)/,ERXQG 3'(% ! **$AUHSHDWV 3335$ ABI3 '1$-& '863 13<5 .&1( 5%0 DOO**$AUHSHDWUHJLRQV 1.; NEDURXQGWKH766 9$9 ;* .&1$% 4 .&1$ 51) 0<20 3 355/ 355T4 &'$ $'5% 2 '&'& 711, 8*7$ 3+263+2 )(=) *1*7 ORJ QXPEHURIUHSHDWV A571 0 13<5 /2;+' /,3, 5$; 0 40 60 20 GLVWDQFHIURP766 NE D TCGA 35$0( 5 code cancer normal tumor LAML Acute Myeloid Leukemia 0 151 ORJ )3.0 ACC AdrenocoƌƟcal carcinoma 0 79 0 BLCA Bladder Urothelial Carcinoma 19 411 LGG Brain Lower Grade Glioma 0 529 COAD Colon adenocarcinoma 41 464 5%0 ESCA Esophageal carcinoma 11 162 GBM Glioblastoma muůƟforme 5 168 5 KICH Kidney Chromophobe 24 65 KIRP Kidney renal papillary cell carcinoma 32 289 LIHC Liver hepatocellular carcinoma 50 374 ORJ )3.0 LUAD Lung adenocarcinoma 59 526 0 LUSC Lung squamous cell carcinoma 49 501 DLBC Lymphoid Neoplasm Diīuse Large B-cell Lym 042 /,3, MESO Mesothelioma 0 1 OV Ovarian serous cystadenocarcinoma 0 379 PAAD PancreĂƟc adenocarcinoma 4 178 5 PCPG Pheochromocytoma and Paraganglioma 3 150 PRAD Prostate adenocarcinoma 52 499 READ Rectum adenocarcinoma 1 6 ORJ )3.0 SARC Sarcoma 0 35 0 SKCM Skin Cutaneous Melanoma 1 471 STAD Stomach adenocarcinoma 32 375 /2;+' WXPRU TGCT dĞƐƟĐƵůĂƌ'ĞƌŵĞůůdƵŵŽƌƐ 0156 THCA Thyroid carcinoma 58 510 QRUPDO UCS Uterine Carcinosarcoma 0 56 5 ORJ )3.0 0 / / 29 $&& 8&6 /** *%0 -

Complexin-1 and Foxp1 Expression Changes Are Novel Brain Effects of Alpha-Synuclein Pathology
Mol Neurobiol DOI 10.1007/s12035-014-8844-0 Complexin-1 and Foxp1 Expression Changes Are Novel Brain Effects of Alpha-Synuclein Pathology Suzana Gispert & Alexander Kurz & Nadine Brehm & Katrin Rau & Michael Walter & Olaf Riess & Georg Auburger Received: 15 April 2014 /Accepted: 31 July 2014 # The Author(s) 2014. This article is published with open access at Springerlink.com Abstract As the second most frequent neurodegenerative Introduction disorder of the aging population, Parkinson’s disease (PD) is characterized by progressive deficits in spontaneous move- Parkinson’s disease (PD) is the second most frequent age- ment, atrophy of dopaminergic midbrain neurons and aggre- associated brain degeneration disorder, affecting about 1 % gation of the protein alpha-synuclein (SNCA). To elucidate of the population over 65 years of age. The PD-specific molecular events before irreversible cell death, we studied progressive movement deficit is mostly due to the severe synucleinopathy-induced expression changes in mouse brain affliction and cell death of midbrain nigrostriatal dopaminer- and identified 49 midbrain/brainstem-specific transcriptional gic neurons [1]. Surviving neurons in vulnerable regions dysregulations. In particular complexin-1 (Cplx1), Rabl2a and exhibit aggregates predominantly consisting of the protein 14-3-3epsilon (Ywhae) downregulation, as well as upregula- alpha-synuclein, which are visualized as Lewy neurites and tion of the midbrain-specific factor forkhead box P1 (Foxp1) Lewy bodies, both in sporadic late-onset and most familial and of Rabgef1, were interesting as early mRNA level effects early onset PD variants [2]. of alpha-synuclein triggered pathology. The protein levels of Autosomal dominant PD with early clinical manifestation complexin-1 were elevated in midbrain/brainstem tissue of was observed in rare families, leading to the identification of mice with A53T-SNCA overexpression and of mice with alpha-synuclein (SNCA) protein missense mutations such as SNCA-knockout. -

Transcriptomics Uncovers Substantial Variability Associated with Alterations in Manufacturing Processes of Macrophage Cell Therapy Products Olga L
www.nature.com/scientificreports OPEN Transcriptomics uncovers substantial variability associated with alterations in manufacturing processes of macrophage cell therapy products Olga L. Gurvich1,3, Katja A. Puttonen1,3, Aubrey Bailey1, Anssi Kailaanmäki1, Vita Skirdenko1, Minna Sivonen1, Sanna Pietikäinen1, Nigel R. Parker2, Seppo Ylä‑Herttuala2 & Tuija Kekarainen1* Gene expression plasticity is central for macrophages’ timely responses to cues from the microenvironment permitting phenotypic adaptation from pro‑infammatory (M1) to wound healing and tissue‑regenerative (M2, with several subclasses). Regulatory macrophages are a distinct macrophage type, possessing immunoregulatory, anti‑infammatory, and angiogenic properties. Due to these features, regulatory macrophages are considered as a potential cell therapy product to treat clinical conditions, e.g., non‑healing diabetic foot ulcers. In this study we characterized two diferently manufactured clinically relevant regulatory macrophages, programmable cells of monocytic origin and comparator macrophages (M1, M2a and M0) using fow‑cytometry, RT‑qPCR, phagocytosis and secretome measurements, and RNA‑Seq. We demonstrate that conventional phenotyping had a limited potential to discriminate diferent types of macrophages which was ameliorated when global transcriptome characterization by RNA‑Seq was employed. Using this approach we confrmed that macrophage manufacturing processes can result in a highly reproducible cell phenotype. At the same time, minor changes introduced in manufacturing -

Investigating Contributions of Trisomy 21 in Down Syndrome to Alzheimer Disease Phenotypes in a Novel Mouse Cross
Investigating contributions of trisomy 21 in Down syndrome to Alzheimer disease phenotypes in a novel mouse cross A thesis presented in partial fulfilment of the requirements for the degree of Doctor of Philosophy from University College London Xun Yu Choong Department of Neurodegenerative Disease Institute of Neurology University College London 2016 1 Declaration I, Xun Yu Choong, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 Acknowledgements The work done in this project was only possible with the support of numerous friends and colleagues, with whom I have grown an incredible amount. Foremost thanks go to Prof. Elizabeth Fisher and Dr. Frances Wiseman, who have been inexhaustibly dedicated supervisors and inspirational figures. Special mention also goes to two unofficial mentors who have looked out for me throughout the PhD, Dr. Karen Cleverley and Dr. Sarah Mizielinska. The Fisher and Isaacs groups have been a joy to work with and have always unhesitatingly offered time and help. Thank you to the Down syndrome group: Olivia Sheppard, Dr. Toby Collins, Dr. Suzanna Noy, Amy Nick, Laura Pulford, Justin Tosh, Matthew Rickman; other members of Lizzy’s group: Dr. Anny Devoy, Dr. Rosie Bunton-Stasyshyn, Dr. Rachele Saccon, Julian Pietrzyk, Dr. Beverley Burke, Heesoon Park, Julian Jaeger; the Isaacs group: Dr. Adrian Isaacs, Dr. Emma Clayton, Dr. Roberto Simone, Charlotte Ridler, Frances Norona. The PhD office has also been a second home in more ways than one, thanks to: Angelos Armen, Dr. -

Journal of Down Syndrome & Chromosome Abnormalities
e & Ch m rom ro o d s n o y m S e n A Journal of Down Syndrome & w Lauricella SA et al., J Down Syndr Chr Abnorm b o n D o f r m 2016, 2:2 o l ISSN: 2472-1115a a l i n t r i e u s o J Chromosome Abnormalities DOI: 10.4172/2472-1115.1000110 Case Report Open Access A Rare Complex Structural Chromosomal Anomaly in Mosaic Due to the Instability of a Derivative Chromosome 18 in a Female Infertile Patient Salvatrice A Lauricella1, Martina Busè2*, Simona Cavani3, Helenia C Cuttaia1, Michela Malacarne3, Marcella V Mazara1, Mauro Pierluigi3 and Maria Piccione2 1Laboratory of Medical Cytogenetic, AOOR Villa Sofia-Cervello, Palermo, Italy 2Department of Sciences for Health Promotion and Mother and Child Care “Giuseppe D’Alessandro”, University of Palermo, Palermo, Italy 3S.C. Laboratory of Human Genetics, E.O. Galliera Hospital, Genova, Italy *Corresponding author: Martina Busè, via Alfonso Giordano, 390127 Palermo (PA), Tel: 0039 091 23869601; E-mail: [email protected] Received date: May 11, 2016; Accepted date: June 23, 2016; Published date: June 30, 2016 Copyright: © 2016 Lauricella SA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited Abstract Mosaicism in association with derivative chromosomes is a well-known fact. Derivative chromosomes often tend to rearrange and/or be reduced in size during karyotype evolution, because of their instability. This can determine the disappearance of a derivative chromosome at least in the most frequently studied tissue, the peripheral blood.