Identification of the Genomic Breakpoints of a Novel Chromosome Translocation in Myelodysplastic Syndrome
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Genetic and Pharmacological Approaches to Preventing Neurodegeneration
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2012 Genetic and Pharmacological Approaches to Preventing Neurodegeneration Marco Boccitto University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Neuroscience and Neurobiology Commons Recommended Citation Boccitto, Marco, "Genetic and Pharmacological Approaches to Preventing Neurodegeneration" (2012). Publicly Accessible Penn Dissertations. 494. https://repository.upenn.edu/edissertations/494 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/494 For more information, please contact [email protected]. Genetic and Pharmacological Approaches to Preventing Neurodegeneration Abstract The Insulin/Insulin-like Growth Factor 1 Signaling (IIS) pathway was first identified as a major modifier of aging in C.elegans. It has since become clear that the ability of this pathway to modify aging is phylogenetically conserved. Aging is a major risk factor for a variety of neurodegenerative diseases including the motor neuron disease, Amyotrophic Lateral Sclerosis (ALS). This raises the possibility that the IIS pathway might have therapeutic potential to modify the disease progression of ALS. In a C. elegans model of ALS we found that decreased IIS had a beneficial effect on ALS pathology in this model. This beneficial effect was dependent on activation of the transcription factor daf-16. To further validate IIS as a potential therapeutic target for treatment of ALS, manipulations of IIS in mammalian cells were investigated for neuroprotective activity. Genetic manipulations that increase the activity of the mammalian ortholog of daf-16, FOXO3, were found to be neuroprotective in a series of in vitro models of ALS toxicity. -

RET Gene Fusions in Malignancies of the Thyroid and Other Tissues
G C A T T A C G G C A T genes Review RET Gene Fusions in Malignancies of the Thyroid and Other Tissues Massimo Santoro 1,*, Marialuisa Moccia 1, Giorgia Federico 1 and Francesca Carlomagno 1,2 1 Department of Molecular Medicine and Medical Biotechnology, University of Naples “Federico II”, 80131 Naples, Italy; [email protected] (M.M.); [email protected] (G.F.); [email protected] (F.C.) 2 Institute of Endocrinology and Experimental Oncology of the CNR, 80131 Naples, Italy * Correspondence: [email protected] Received: 10 March 2020; Accepted: 12 April 2020; Published: 15 April 2020 Abstract: Following the identification of the BCR-ABL1 (Breakpoint Cluster Region-ABelson murine Leukemia) fusion in chronic myelogenous leukemia, gene fusions generating chimeric oncoproteins have been recognized as common genomic structural variations in human malignancies. This is, in particular, a frequent mechanism in the oncogenic conversion of protein kinases. Gene fusion was the first mechanism identified for the oncogenic activation of the receptor tyrosine kinase RET (REarranged during Transfection), initially discovered in papillary thyroid carcinoma (PTC). More recently, the advent of highly sensitive massive parallel (next generation sequencing, NGS) sequencing of tumor DNA or cell-free (cfDNA) circulating tumor DNA, allowed for the detection of RET fusions in many other solid and hematopoietic malignancies. This review summarizes the role of RET fusions in the pathogenesis of human cancer. Keywords: kinase; tyrosine kinase inhibitor; targeted therapy; thyroid cancer 1. The RET Receptor RET (REarranged during Transfection) was initially isolated as a rearranged oncoprotein upon the transfection of a human lymphoma DNA [1]. -

Apoptotic Genes As Potential Markers of Metastatic Phenotype in Human Osteosarcoma Cell Lines
17-31 10/12/07 14:53 Page 17 INTERNATIONAL JOURNAL OF ONCOLOGY 32: 17-31, 2008 17 Apoptotic genes as potential markers of metastatic phenotype in human osteosarcoma cell lines CINZIA ZUCCHINI1, ANNA ROCCHI2, MARIA CRISTINA MANARA2, PAOLA DE SANCTIS1, CRISTINA CAPANNI3, MICHELE BIANCHINI1, PAOLO CARINCI1, KATIA SCOTLANDI2 and LUISA VALVASSORI1 1Dipartimento di Istologia, Embriologia e Biologia Applicata, Università di Bologna, Via Belmeloro 8, 40126 Bologna; 2Laboratorio di Ricerca Oncologica, Istituti Ortopedici Rizzoli; 3IGM-CNR, Unit of Bologna, c/o Istituti Ortopedici Rizzoli, Via di Barbiano 1/10, 40136 Bologna, Italy Received May 29, 2007; Accepted July 19, 2007 Abstract. Metastasis is the most frequent cause of death among malignant primitive bone tumor, usually developing in children patients with osteosarcoma. We have previously demonstrated and adolescents, with a high tendency to metastasize (2). in independent experiments that the forced expression of Metastases in osteosarcoma patients spread through peripheral L/B/K ALP and CD99 in U-2 OS osteosarcoma cell lines blood very early and colonize primarily the lung, and later markedly reduces the metastatic ability of these cancer cells. other skeleton districts (3). Since disseminated hidden micro- This behavior makes these cell lines a useful model to assess metastases are present in 80-90% of OS patients at the time the intersection of multiple and independent gene expression of diagnosis, the identification of markers of invasiveness signatures concerning the biological problem of dissemination. and metastasis forms a target of paramount importance in With the aim to characterize a common transcriptional profile planning the treatment of osteosarcoma lesions and enhancing reflecting the essential features of metastatic behavior, we the prognosis. -

Molecular Evolutionary Analysis of Plastid Genomes in Nonphotosynthetic Angiosperms and Cancer Cell Lines
The Pennsylvania State University The Graduate School Department or Biology MOLECULAR EVOLUTIONARY ANALYSIS OF PLASTID GENOMES IN NONPHOTOSYNTHETIC ANGIOSPERMS AND CANCER CELL LINES A Dissertation in Biology by Yan Zhang 2012 Yan Zhang Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Dec 2012 The Dissertation of Yan Zhang was reviewed and approved* by the following: Schaeffer, Stephen W. Professor of Biology Chair of Committee Ma, Hong Professor of Biology Altman, Naomi Professor of Statistics dePamphilis, Claude W Professor of Biology Dissertation Adviser Douglas Cavener Professor of Biology Head of Department of Biology *Signatures are on file in the Graduate School iii ABSTRACT This thesis explores the application of evolutionary theory and methods in understanding the plastid genome of nonphotosynthetic parasitic plants and role of mutations in tumor proliferations. We explore plastid genome evolution in parasitic angiosperms lineages that have given up the primary function of plastid genome – photosynthesis. Genome structure, gene contents, and evolutionary dynamics were analyzed and compared in both independent and related parasitic plant lineages. Our studies revealed striking similarities in changes of gene content and evolutionary dynamics with the loss of photosynthetic ability in independent nonphotosynthetic plant lineages. Evolutionary analysis suggests accelerated evolution in the plastid genome of the nonphotosynthetic plants. This thesis also explores the application of phylogenetic and evolutionary analysis in cancer biology. Although cancer has often been likened to Darwinian process, very little application of molecular evolutionary analysis has been seen in cancer biology research. In our study, phylogenetic approaches were used to explore the relationship of several hundred established cancer cell lines based on multiple sequence alignments constructed with variant codons and residues across 494 and 523 genes. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Mouse Hnrnph3 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Hnrnph3 Knockout Project (CRISPR/Cas9) Objective: To create a Hnrnph3 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Hnrnph3 gene (NCBI Reference Sequence: NM_001079824 ; Ensembl: ENSMUSG00000020069 ) is located on Mouse chromosome 10. 10 exons are identified, with the ATG start codon in exon 2 and the TGA stop codon in exon 10 (Transcript: ENSMUST00000020263). Exon 2~3 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 2 starts from the coding region. Exon 2~3 covers 24.18% of the coding region. The size of effective KO region: ~768 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 10 Legends Exon of mouse Hnrnph3 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 534 bp section downstream of Exon 3 is aligned with itself to determine if there are tandem repeats. -

Supplementary Tables S1-S3
Supplementary Table S1: Real time RT-PCR primers COX-2 Forward 5’- CCACTTCAAGGGAGTCTGGA -3’ Reverse 5’- AAGGGCCCTGGTGTAGTAGG -3’ Wnt5a Forward 5’- TGAATAACCCTGTTCAGATGTCA -3’ Reverse 5’- TGTACTGCATGTGGTCCTGA -3’ Spp1 Forward 5'- GACCCATCTCAGAAGCAGAA -3' Reverse 5'- TTCGTCAGATTCATCCGAGT -3' CUGBP2 Forward 5’- ATGCAACAGCTCAACACTGC -3’ Reverse 5’- CAGCGTTGCCAGATTCTGTA -3’ Supplementary Table S2: Genes synergistically regulated by oncogenic Ras and TGF-β AU-rich probe_id Gene Name Gene Symbol element Fold change RasV12 + TGF-β RasV12 TGF-β 1368519_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 42.22 5.53 75.28 1373000_at sushi-repeat-containing protein, X-linked 2 (predicted) Srpx2 19.24 25.59 73.63 1383486_at Transcribed locus --- ARE 5.93 27.94 52.85 1367581_a_at secreted phosphoprotein 1 Spp1 2.46 19.28 49.76 1368359_a_at VGF nerve growth factor inducible Vgf 3.11 4.61 48.10 1392618_at Transcribed locus --- ARE 3.48 24.30 45.76 1398302_at prolactin-like protein F Prlpf ARE 1.39 3.29 45.23 1392264_s_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 24.92 3.67 40.09 1391022_at laminin, beta 3 Lamb3 2.13 3.31 38.15 1384605_at Transcribed locus --- 2.94 14.57 37.91 1367973_at chemokine (C-C motif) ligand 2 Ccl2 ARE 5.47 17.28 37.90 1369249_at progressive ankylosis homolog (mouse) Ank ARE 3.12 8.33 33.58 1398479_at ryanodine receptor 3 Ryr3 ARE 1.42 9.28 29.65 1371194_at tumor necrosis factor alpha induced protein 6 Tnfaip6 ARE 2.95 7.90 29.24 1386344_at Progressive ankylosis homolog (mouse) -

Endocrine System Local Gene Expression
Copyright 2008 By Nathan G. Salomonis ii Acknowledgments Publication Reprints The text in chapter 2 of this dissertation contains a reprint of materials as it appears in: Salomonis N, Hanspers K, Zambon AC, Vranizan K, Lawlor SC, Dahlquist KD, Doniger SW, Stuart J, Conklin BR, Pico AR. GenMAPP 2: new features and resources for pathway analysis. BMC Bioinformatics. 2007 Jun 24;8:218. The co-authors listed in this publication co-wrote the manuscript (AP and KH) and provided critical feedback (see detailed contributions at the end of chapter 2). The text in chapter 3 of this dissertation contains a reprint of materials as it appears in: Salomonis N, Cotte N, Zambon AC, Pollard KS, Vranizan K, Doniger SW, Dolganov G, Conklin BR. Identifying genetic networks underlying myometrial transition to labor. Genome Biol. 2005;6(2):R12. Epub 2005 Jan 28. The co-authors listed in this publication developed the hierarchical clustering method (KP), co-designed the study (NC, AZ, BC), provided statistical guidance (KV), co- contributed to GenMAPP 2.0 (SD) and performed quantitative mRNA analyses (GD). The text of this dissertation contains a reproduction of a figure from: Yeo G, Holste D, Kreiman G, Burge CB. Variation in alternative splicing across human tissues. Genome Biol. 2004;5(10):R74. Epub 2004 Sep 13. The reproduction was taken without permission (chapter 1), figure 1.3. iii Personal Acknowledgments The achievements of this doctoral degree are to a large degree possible due to the contribution, feedback and support of many individuals. To all of you that helped, I am extremely grateful for your support. -
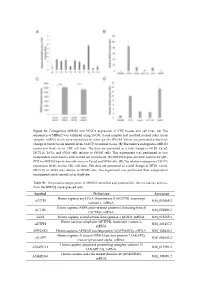
Figure S1. Endogenous MIR45
Figure S1. Endogenous MIR452 and VEGFA expression in CRC tissues and cell lines. (A) The expression of MIR452 was validated using 10 CRC tissue samples and matched normal colon tissue samples. miRNA levels were normalized to colon-specific RNU48. Values are presented as the fold- change in tumor tissue relative levels (ΔΔCT) to normal tissue. (B) The relative endogenous MIR452 expression levels in six CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, LoVo, and SW48 cells relative to SW480 cells. This experiment was performed as two independent experiments, each carried out in triplicate. (C) MIR452 expression level analysis by qRT- PCR for MIR452 transfection efficiency in Caco2 and SW48 cells. (D) The relative endogenous VEGFA expression levels in five CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, or SW48 cells relative to SW480 cells. This experiment was performed three independent experiments, each carried out in duplicate. Table S1. The putative target genes of MIR452 identified and predicted by the microarray analysis from the MIR452 overexpressed cells. Symbol Definition Accession Homo sapiens acyl-CoA thioesterase 8 (ACOT8), transcript ACOT8 NM_005469.2 variant 1, mRNA. Homo sapiens ARP6 actin-related protein 6 homolog (yeast) ACTR6 NM_022496.3 (ACTR6), mRNA. ADI1 Homo sapiens acireductone dioxygenase 1 (ADI1), mRNA. NM_018269.1 Homo sapiens aftiphilin (AFTPH), transcript variant 1, AFTPH NM_203437.2 mRNA. AHNAK2 Homo sapiens AHNAK nucleoprotein 2 (AHNAK2), mRNA. NM_138420.2 Homo sapiens A kinase (PRKA) anchor protein 7 (AKAP7), AKAP7 NM_004842.2 transcript variant alpha, mRNA. Homo sapiens anaphase promoting complex subunit 13 ANAPC13 NM_015391.2 (ANAPC13), mRNA. -

(12) Patent Application Publication (10) Pub. No.: US 2003/0170678A1 Tanzi Et Al
US 2003.017O678A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0170678A1 Tanzi et al. (43) Pub. Date: Sep. 11, 2003 (54) GENETIC MARKERS FOR ALZHEIMER'S Related U.S. Application Data DISEASE AND METHODS USING THE SAME (60) Provisional application No. 60/348,065, filed on Oct. (75) Inventors: Rudolph E. Tanzi, Hull, MA (US); 25, 2001. Provisional application No. 60/336,983, Kenneth David Becker, San Diego, CA filed on Nov. 2, 2001. (US); Gonul Velicelebi, San Diego, CA (US); Kathryn J. Elliott, San Diego, Publication Classification CA (US); Xin Wang, San Diego, CA (US); Lars Bertram, Brighton, MA (51) Int. C.7 - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - C12O 1/68 (US); Aleister J. Saunders, (52) U.S. Cl. .................................................................. 435/6 Philadelphia, PA (US); Deborah Lynne Blacker, Newton, MA (US) (57) ABSTRACT Correspondence Address: HELLER EHRMAN WHITE & MCAULIFFE LLP Genetic markers associated with Alzheimer's disease are 4350 LA JOLLAVILLAGE DRIVE provided. Also provided are methods of determining the 7TH FLOOR presence or absence in a Subject of one or more polymor SAN DIEGO, CA 92122-1246 (US) phisms associated with Alzheimer's disease and methods of determining the level of risk for Alzheimer's disease in a (73) Assignee: Neurogenetics, Inc. Subject. Further provided are nucleic acid compositions and kits for use in determining the presence or absence in a (21) Appl. No.: 10/281,456 Subject of one or more polymorphisms associated with Alzheimer's disease and kits for determining the level of (22) Filed: Oct. 25, 2002 risk for Alzheimer's disease in a Subject. Patent Application Publication Sep. -

Translational Regulation of 5' Terminal Oligopyrimidine Tract (TOP) Mrnas by Insulin
Markou et al. BMC Genomics 2010, 11:343 http://www.biomedcentral.com/1471-2164/11/343 RESEARCH ARTICLE Open Access RegulationResearch article of the cardiomyocyte transcriptome vs translatome by endothelin-1 and insulin: translational regulation of 5' terminal oligopyrimidine tract (TOP) mRNAs by insulin Thomais Markou, Andrew K Marshall, Timothy E Cullingford, El L Tham, Peter H Sugden and Angela Clerk* Abstract Background: Changes in cellular phenotype result from underlying changes in mRNA transcription and translation. Endothelin-1 stimulates cardiomyocyte hypertrophy with associated changes in mRNA/protein expression and an increase in the rate of protein synthesis. Insulin also increases the rate of translation but does not promote overt cardiomyocyte hypertrophy. One mechanism of translational regulation is through 5' terminal oligopyrimidine tracts (TOPs) that, in response to growth stimuli, promote mRNA recruitment to polysomes for increased translation. TOP mRNAs include those encoding ribosomal proteins, but the full panoply remains to be established. Here, we used microarrays to compare the effects of endothelin-1 and insulin on the global transcriptome of neonatal rat cardiomyocytes, and on mRNA recruitment to polysomes (i.e. the translatome). Results: Globally, endothelin-1 and insulin (1 h) promoted >1.5-fold significant (false discovery rate < 0.05) changes in expression of 341 and 38 RNAs, respectively. For these transcripts with this level of change there was little evidence of translational regulation. However, 1336 and 712 RNAs had >1.25-fold significant changes in expression in total and/or polysomal RNA induced by endothelin-1 or insulin, respectively, of which ~35% of endothelin-1-responsive and ~56% of insulin-responsive transcripts were translationally regulated. -

Genomic Analyses Reveal Recurrent Mutations in Epigenetic Modifiers
ARTICLE Received 6 Jul 2015 | Accepted 25 Aug 2015 | Published 29 Sep 2015 DOI: 10.1038/ncomms9470 OPEN Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK–STAT pathway in Se´zary syndrome Mark J. Kiel1,*, Anagh A. Sahasrabuddhe1,*,w, Delphine C.M. Rolland2,*, Thirunavukkarasu Velusamy1,*, Fuzon Chung1, Matthew Schaller1, Nathanael G. Bailey1, Bryan L. Betz1, Roberto N. Miranda3, Pierluigi Porcu4, John C. Byrd4, L. Jeffrey Medeiros3, Steven L. Kunkel1, David W. Bahler5, Megan S. Lim2 & Kojo S.J. Elenitoba-Johnson2,6 Se´zary syndrome (SS) is an aggressive leukaemia of mature T cells with poor prognosis and limited options for targeted therapies. The comprehensive genetic alterations underlying the pathogenesis of SS are unknown. Here we integrate whole-genome sequencing (n ¼ 6), whole-exome sequencing (n ¼ 66) and array comparative genomic hybridization-based copy- number analysis (n ¼ 80) of primary SS samples. We identify previously unknown recurrent loss-of-function aberrations targeting members of the chromatin remodelling/histone mod- ification and trithorax families, including ARID1A in which functional loss from nonsense and frameshift mutations and/or targeted deletions is observed in 40.3% of SS genomes. We also identify recurrent gain-of-function mutations targeting PLCG1 (9%) and JAK1, JAK3, STAT3 and STAT5B (JAK/STAT total B11%). Functional studies reveal sensitivity of JAK1-mutated primary SS cells to JAK inhibitor treatment. These results highlight the complex genomic landscape of SS and a role for inhibition of JAK/STAT pathways for the treatment of SS. 1 Department of Pathology, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA.