Thesis Rests with Its Author
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Vascular Plants of Massachusetts
The Vascular Plants of Massachusetts: The Vascular Plants of Massachusetts: A County Checklist • First Revision Melissa Dow Cullina, Bryan Connolly, Bruce Sorrie and Paul Somers Somers Bruce Sorrie and Paul Connolly, Bryan Cullina, Melissa Dow Revision • First A County Checklist Plants of Massachusetts: Vascular The A County Checklist First Revision Melissa Dow Cullina, Bryan Connolly, Bruce Sorrie and Paul Somers Massachusetts Natural Heritage & Endangered Species Program Massachusetts Division of Fisheries and Wildlife Natural Heritage & Endangered Species Program The Natural Heritage & Endangered Species Program (NHESP), part of the Massachusetts Division of Fisheries and Wildlife, is one of the programs forming the Natural Heritage network. NHESP is responsible for the conservation and protection of hundreds of species that are not hunted, fished, trapped, or commercially harvested in the state. The Program's highest priority is protecting the 176 species of vertebrate and invertebrate animals and 259 species of native plants that are officially listed as Endangered, Threatened or of Special Concern in Massachusetts. Endangered species conservation in Massachusetts depends on you! A major source of funding for the protection of rare and endangered species comes from voluntary donations on state income tax forms. Contributions go to the Natural Heritage & Endangered Species Fund, which provides a portion of the operating budget for the Natural Heritage & Endangered Species Program. NHESP protects rare species through biological inventory, -

Nursery Price List
Lincoln-Oakes Nurseries 3310 University Drive • Bismarck, ND 58504 Nursery Seed Price List 701-223-8575 • [email protected] The following seed is in stock or will be collected and available for 2010 or spring 2011 PENDING CROP, all climatic zone 3/4 collections from established plants in North Dakota except where noted. Acer ginnala - 18.00/lb d.w Cornus racemosa - 19.00/lb Amur Maple Gray dogwood Acer tataricum - 15.00/lb d.w Cornus alternifolia - 21.00/lb Tatarian Maple Pagoda dogwood Aesculus glabra (ND, NE) - 3.95/lb Cornus stolonifera (sericea) - 30.00/lb Ohio Buckeye – collected from large well performing Redosier dogwood Trees in upper midwest Amorpha canescens - 90.00/lb Leadplant 7.50/oz Amorpha fruiticosa - 10.50/lb False Indigo – native wetland restoration shrub Aronia melanocarpa ‘McKenzie” - 52.00/lb Black chokeberry - taller form reaching 6-8 ft in height, glossy foliage, heavy fruit production, Corylus cornuta (partial husks) - 16.00/lb NRCS release Beaked hazelnut/Native hazelnut (Inquire) Caragana arborescens - 16.00/lb Cotoneaster integerrimus ‘Centennial’ - 32.00/lb Siberian peashrub European cotoneaster – NRCS release, 6-10’ in height, bright red fruit Celastrus scandens (true) (Inquire) - 58.00/lb American bittersweet, no other contaminating species in area Crataegus crus-galli - 22.00/lb Cockspur hawthorn, seed from inermis Crataegus mollis ‘Homestead’ arnoldiana-24.00/lb Arnold hawthorn – NRCS release Crataegus mollis - 19.50/lb Downy hawthorn Elaeagnus angustifolia - 9.00/lb Russian olive Elaeagnus commutata -

Gray Dogwood Cornus Racemosa ILLINOIS RANGE Leaves and Flowers
gray dogwood Cornus racemosa Kingdom: Plantae FEATURES Division: Magnoliophyta Gray dogwood is also known as gray-stemmed Class: Magnoliopsida dogwood or panicled dogwood. This shrub may Order: Cornales attain a height of 10 feet. Its opposite, gray-green leaves are simple and entire. Its bark is smooth and Family: Cornaceae light gray. The slender, gray twigs have dark pith. ILLINOIS STATUS Small, white flowers develop in clusters. The white berries (drupes) produced, each about one-fourth common, native inch in diameter, have red stems. BEHAVIORS Gray dogwood may be found statewide in Illinois. It grows in moist woods, upland woods, dry to wet prairies and along roadsides. Flowers are produced from May through July. Gray dogwood plants grow in clusters that give the appearance of a mound. Several species of songbirds take advantage of the closely-spaced twigs and camouflage of the leaves to build their nest in gray dogwood thickets. The leaves, buds, twigs and fruit of this plant are used as food by some wildlife species. Seeds are dispersed in the waste material of birds and mammals that eat the fruit. Leaves turn red or purple in the fall of the year. ILLINOIS RANGE leaves and flowers © Illinois Department of Natural Resources. 2021. Biodiversity of Illinois. Unless otherwise noted, photos and images © Illinois Department of Natural Resources. leaves and flowers Aquatic Habitats bottomland forests; wet prairies and fens Woodland Habitats bottomland forests; coniferous forests; southern Illinois lowlands; upland deciduous forests Prairie and Edge Habitats black soil prairie © Illinois Department of Natural Resources. 2021. Biodiversity of Illinois. Unless otherwise noted, photos and images © Illinois Department of Natural Resources.. -

Selecting Trees and Shrubs in Windbreaks
Fall 2012 Selecting Trees and Shrubs in Windbreaks Windbreaks are plantings of single or multiple rows of trees or shrubs that are established for one or more environmental purposes. Purposes of windbreaks include: wind protection, controlling blowing and drifting snow, wildlife habitat establishment, energy saving, living screens, odor abatement and more. The effectiveness of a windbreak depends on suitable tree and shrub selection as well as planting density and spacing. This fact sheet offers information on tree and shrub species to consider in Minnesota windbreak plantings and a list of resources for technical and financial assistance. Tree and Shrub Selection: Windbreak tree and shrub selection is extremely important to insure an effective, long lasting planting. Plants need to be winter hardy and should have a positive history of plantings in the area suitable for the site and soils. Select multiple species of trees and shrubs, so if there is a failure in a row the windbreak is still effective for the purposes. A mix of deciduous and coniferous plants is recommended and should be based on the purpose of the planting. Many plants can offer potential income benefits such as edible, decorative, crafts, medicinal and specialty woods. As appropriate, first consider using native plants. Landowners should consult with area or County Soil and Water Conservation Districts/National Resources Conservation Service, Department of Natural Resource and Extension staff to get a recommended list of plants suitable for the area. Density: How dense -

Cornaceae – Dogwood Family
CORNACEAE – DOGWOOD FAMILY Plant: shrubs and small trees (possibly herbs elsewhere) Stem: twigs with white or brown pith Root: Leaves: mostly deciduous but some evergreen; mostly not toothed but may be wavy, opposite or less often alternate; no stipules Flowers: mostly perfect locally (or dioecious elsewhere); sometimes with large petal-like bracts; small, mostly 4-5 sepals, often just teeth or minute; mostly 4-5 petals or rarely none; 4-10 (15) stamens; ovary inferior, usually 2 carpels, usually 1 style Fruit: drupe or berry, oily, 1-2 seeded Other: widespread in tropical and temperate regions; locally the dogwoods; some put Nyssa (sour gum genus) in this family instead of in the Nyssaceae; Dicotyledons Group Genera: 65+ species; locally Cornus (dogwood) – Some would put Nyssa and relatives here instead of the family Nyssaeae WARNING – family descriptions are only a layman’s guide and should not be used as definitive Flower Morphology in the Cornaceae (Dogwood Family) Examples of genus Cornus Silky Dogwood Cornus amomum Mill. Flowering Dogwood Cornus florida L. Roughleaf Dogwood Cornus drummondii C.A. Mey. Bunchberry Dogwood [Dwarf Cornel] Cornus canadensis L. CORNACEAE – DOGWOOD FAMILY Alternate Leaf [Pagoda] Dogwood; Cornus alternifolia L. f. Silky Dogwood; Cornus amomum Mill. Bunchberry Dogwood [Dwarf Cornel]; Cornus canadensis L. Roughleaf Dogwood; Cornus drummondii C.A. Mey. Flowering Dogwood; Cornus florida L. Kousa Dogwood; Cornus kousa Hance (Introduced) Gray [Panicled] Dogwood; Cornus racemosa Lam. Red Ossier; Cornus sericea -

Cornus, Cornaceae) Dans L'herbier De Strasbourg (STR
1 Les Cornouillers ( Cornus , Cornaceae) dans l’Herbier de Strasbourg (STR) Yvan Brahy et Michel Hoff Avec la collaboration de Françoise Deluzarche, Frédéric Tournay, Gisèle Haan-Archipof et Claudine Bertin-Charbonnier Herbier de l’Université de Strasbourg 2 3 Plan Introduction 1. Etude nomenclaturale et taxonomique des Cornus des Herbiers de Strasbourg. 1. Cornus alba C. Linnaeus 1.2. Cornus alternifolia C. Linnaeus f. 1.3. Cornus amomum Mill. 1.4. Cornus asperifolia Michaux. 1.5. Cornus canadensis C. Linnaeus 1.6. Cornus capitata Wall. 1.7. Cornus disciflora Moc. & Sessé ex D.C. 1.8. Cornus excelsa Kunth 1.9. Cornus florida C. Linnaeus 1.10. Cornus foemina Mill. 1.11. Cornus kousa F. Buerger ex Miquel 1.12. Cornus macrophylla Wall. 1.13. Cornus mas C. Linnaeus 1.14. Cornus nuttallii Audubon ex Torr. & A. Gray 1.15. Cornus officinalis Siebold & Zucc. 1.16. Cornus quinquinervis Franch. 1.17. Cornus racemosa Lam. 1.18. Cornus rugosa Lam. 1.19. Cornus sanguinea C. Linnaeus 1.20. Cornus sericea C. Linnaeus 1.21. Cornus suecica C. Linnaeus 1.22. Spécimens indéterminés 2. Analyse de la collection des spécimens de l’Herbier des Cornus 2.1. Introduction 2.2. Les collecteurs 2.3. Les collections 2.4. Les années de collecte 2.5. Les pays et régions de collecte 2.6. Les habitats de collecte 2.7. Les principaux jardins botaniques 2.8. Conclusion Conclusion générale Bibliographie Sites internet Annexes Annexe 1 : Caractères des Cornus Annexe 2 : Arbre de parenté des Cornus Annexe 3 : Illustrations 4 Diagramme du genre Cornus 5 Introduction Le genre Cornus rassemble, selon les auteurs, entre 43 et 46 espèces dans le monde (P. -

Chapter Four: Landscaping with Native Plants a Gardener’S Guide for Missouri Landscaping with Native Plants a Gardener’S Guide for Missouri
Chapter Four: Landscaping with Native Plants A Gardener’s Guide for Missouri Landscaping with Native Plants A Gardener’s Guide for Missouri Introduction Gardening with native plants is becoming the norm rather than the exception in Missouri. The benefits of native landscaping are fueling a gardening movement that says “no” to pesticides and fertilizers and “yes” to biodiversity and creating more sustainable landscapes. Novice and professional gardeners are turning to native landscaping to reduce mainte- nance and promote plant and wildlife conservation. This manual will show you how to use native plants to cre- ate and maintain diverse and beauti- ful spaces. It describes new ways to garden lightly on the earth. Chapter Four: Landscaping with Native Plants provides tools garden- ers need to create and maintain suc- cessful native plant gardens. The information included here provides practical tips and details to ensure successful low-maintenance land- scapes. The previous three chap- ters include Reconstructing Tallgrass Prairies, Rain Gardening, and Native landscapes in the Whitmire Wildflower Garden, Shaw Nature Reserve. Control and Identification of Invasive Species. use of native plants in residential gar- den design, farming, parks, roadsides, and prairie restoration. Miller called his History of Native work “The Prairie Spirit in Landscape Landscaping Design”. One of the earliest practitioners of An early proponent of native landscap- Miller’s ideas was Ossian C. Simonds, ing was Wilhelm Miller who was a landscape architect who worked in appointed head of the University of the Chicago region. In a lecture pre- Illinois extension program in 1912. He sented in 1922, Simonds said, “Nature published a number of papers on the Introduction 3 teaches what to plant. -
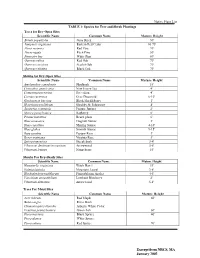
Native Plant List TABLE 1: Species for Tree and Shrub Plantings Trees For
Native Plant List TABLE 1: Species for Tree and Shrub Plantings Trees for Dry-Open Sites Scientific Name Common Name Mature Height Betula populifolia Gray Birch 30' Juniperis virginiana Eastern Red Cedar 10-75' Pinus resinosa Red Pine 70' Pin us rigida Pitch Pine 50' Pinus stro bus White Pine 80' Quercus rubra Red Oak 70' Quercus coccinea Scarlet Oak 70' Quercus velutina Black Oak 70' Shrubs for Dry-Open Sites Scientific Name 'Common Name Mature Height Amelanchier canadensis Shadbush 15' Ceanothus americanus New Jersey Tea 4' Comptonia peregrina Sweetfern 4' Cornus racemosa Gray Dogwood 6-10' Gaylussacia baccata Black Huckleberry l' Hypericum prolificum Shrubby St. Johnswort 4' Juniperus communis Pasture Juniper 2' Myrica pensylvanica Bayberry 6' Prunus maritima Beach plum 6' Rhus aromatica Fragrant Sumac 3' Rhus copallina Shining Sumac 4-10' Rhus glabra Smooth Sumac 9-15' Rosa carolina Pasture Rose 3' Rosa virginiana Virginia Rose 3' Spirea tomentosa Steeplebush 3-4' Viburnum dentatum/recognitum Arrowwood 5-8' Viburnum lentago Nannyberry 15' Shrubs For Dry-Shady Sites Scientific Name Common Name Mature Height Hamamelis wrginiana Witch Hazel 15' Kalmia latifolia Mountain Laurel 3-8' Rhododendron nudiflorum Pinxterbloom Azalea 4-6' Vaccinium angustifolium Lowbush Blueberry 2' Viburnum dentatum Arrowwood 5-8' Trees For Moist Sites Scientific Name Common Name Mature Height Acer rubrum Red Maple 60' Betula nigra River Birch Chamaecyparis thyoides Atlantic White Cedar Fraxinus pennsylvanica Green Ash 60' Picea mariana Black Spruce 40' Picea -

Dogwoods for American Gardens
Agricultural Extension Service The University of Tennessee PB1670 Dogwoods for American Gardens 1 Dogwoods for American Gardens Willard T. Witte, Mark T. Windham, Alan S. Windham, Frank A. Hale, Donna C. Fare and Wayne K. Clatterbuck About the Authors Willard T. Witte, Associate Professor (retired), Dept. of Ornamental Horticulture and Landscape Design, The University of Tennessee Agricultural Experiment Station, Knoxville Mark T. Windham, Professor, Dept. of Entomology and Plant Pathology, The University of Tennessee Agricultural Experiment Station, Knoxville Alan S. Windham, Professor, Dept. of Entomology and Plant Pathology, The University of Tennessee Agricultural Extension Service, Nashville Frank A. Hale, Associate Professor, Dept. of Entomology and Plant Pathology, The University of Tennessee Agricultural Extension Service, Nashville Donna C. Fare, Research Horticulturist, U.S. National Arboretum, Floral & Nursery Plants Research Unit, McMinnville Wayne K. Clatterbuck, Associate Professor, Dept. of Forestry, Wildlife & Fisheries, The University of Tennessee Agricultural Extension Service, Knoxville Acknowledgements The authors acknowledge the contributions of Professors Donald B. Williams, Charles H. Hadden and Harry E. Williams for their original publication entitled “The Flowering Dogwood in Tennessee” (The University of Tennessee Agricultural Extension Service Publication 589, 1969), which was used as a base for this publication. Appreciation is expressed to Hubert P. Conlon, Mark A. Halcomb, Carol J. Reese and Stephen Garton for their peer review of this publication. We also thank Wanda H. Russell for editorial review and Gary R. Dagnan for publication design. Printing of this publication is funded by the USDA Forest Service through a grant with the Na- tional Urban and Community Forestry Advisory Committee. Cover Photo: Wayne K. -

The Use of Regional Phylogenies in Exploring the Structure of Plant Assemblages
The use of regional phylogenies in exploring the structure of plant assemblages Tammy L. Elliott Doctor of Philosophy Department of Biology McGill University Montr´eal, Qu´ebec, Canada 2015-09-015 A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Doctor of Philosophy c Copyright Tammy L. Elliott, 2015 All rights reserved Dedication I dedicate this thesis to my parents, who sadly both left this world much to early. I like to dream that you are both enjoying your time together in a place with no worries, where you can enjoy all of the wonderful things in life. Dad—Although you left us when we were so young, I daily cherish the special times the two of us spent together. The memories of exploring the countryside, visiting neighbours, caring for the pigs and skipping school to fish are always close to my heart. Mom—I miss your strength, interesting perspective (albeit humorously pessimistic), no-nonsense attitude towards life and listening ear. I hope that you are finding ways to enjoy your grandchildren and tend your beautiful gardens. I would like to assure you that yes—one day I will have a full-time job. If Roses grow in Heaven Lord, please pick a bunch for me. Place them in my Mother’s arms and tell her they’re from me. Tell her that I love her and miss her, and when she turns to smile, place a kiss upon her cheek and hold her for awhile Because remembering her is easy, I do it every day, but there’s an ache within my heart that will never go away. -

Chapter Four: Landscaping with Native Plants a Gardeners Guide for Missouri Landscaping with Native Plants a Gardeners Guide for Missouri
Chapter Four: Landscaping with Native Plants A Gardeners Guide for Missouri Landscaping with Native Plants A Gardeners Guide for Missouri Introduction Gardening with native plants is becoming the norm rather than the exception in Missouri. The benefits of native landscaping is fueling a gar- dening movement that says no to pesticides and fertilizers and yes to biodiversity and creates more sus- tainable landscapes. Novice and pro- fessional gardeners are turning to native plants in unprecedented num- bers because these plants can reduce maintenance when used correctly. This manual will show you how to use native plants to reduce mainte- nance, create and maintain diverse and beautiful spaces. It describes new ways to garden lightly on the earth. Chapter four: Landscaping with Native Plants is intended to provide gardeners with the tools they need to create and maintain successful native plant gardens. Whether you are establishing a new garden or expand- published a number of papers on the ing an existing one, the information Native landscapes in the in the Whitmire included here provides practical tips Wildflower Garden, Shaw Nature Reserve. and details to ensure successful low- maintenance landscapes. use of native plants in residential gar- den design, farming, parks, roadsides, History of Native and prairie restoration. Miller called his work “The Prairie Spirit in Landscape Landscaping Design”. One of the earliest practitioners of An early proponent of native landscap- Miller’s ideas was Ossian C. Simonds, a ing was Wilhelm Miller who was landscape architect who worked in the appointed head of the University of Chicago region. In a lecture presented Illinois extension program in 1912. -

A Thesis Entitled Phytoremediation Potential at an Inactive Landfill In
A Thesis Entitled Phytoremediation potential at an inactive landfill in northwest Ohio By Kristopher D. Barnswell Submitted as partial fulfillment of the requirements for the Master of Science degree in Biology __________________________ Advisor: Daryl F. Dwyer __________________________ Graduate School The University of Toledo December 2005 A Thesis Entitled Phytoremediation potential at an inactive landfill in northwest Ohio By Kristopher D. Barnswell Submitted as partial fulfillment of the requirements for the Master of Science degree in Biology ____________________________ __________________________ Jiquan Chen Elliot J. Tramer ____________________________ __________________________ Alison L. Spongberg College of Arts and Sciences ____________________________ Donald J. Stierman The University of Toledo December 2005 Copyright © 2005 This document is copyrighted material. Under copyright law, no parts of this document may be reproduced without the expressed permission of the author. An Abstract for Phytoremediation potential at an inactive landfill in northwest Ohio Kristopher D. Barnswell Submitted as partial fulfillment of the requirements for the Master of Science degree in Biology The University of Toledo December 2005 The King Road Landfill (KRL) received municipal solid waste from 1954 through 1976 that was covered with sandy soil. Concern exists that water may infiltrate through the sandy soil and into the waste thereby producing leachate. Evapotranspiration (ET) covers are a new approach to prevent infiltration of precipitation into landfills. They are less expensive than conventional caps and may perform comparably. The plants chosen to prevent leachate formation can also be used to restore landfills to native habitat. In this study, 191 vascular plant species were identified within 56 plots (each 167 m2) at the KRL and questioned for their appropriateness in creating an ET cover and restoring native habitat.