Microarray Analysis of Iris Gene Expression in Mice with Mutations Influencing Pigmentation
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
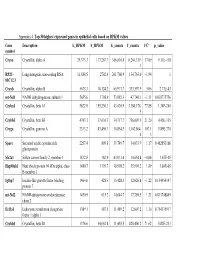
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

CRYGB Rabbit Pab
Leader in Biomolecular Solutions for Life Science CRYGB Rabbit pAb Catalog No.: A14569 Basic Information Background Catalog No. Crystallins are separated into two classes: taxon-specific, or enzyme, and ubiquitous. A14569 The latter class constitutes the major proteins of vertebrate eye lens and maintains the transparency and refractive index of the lens. Since lens central fiber cells lose their Observed MW nuclei during development, these crystallins are made and then retained throughout 21kDa life, making them extremely stable proteins. Mammalian lens crystallins are divided into alpha, beta, and gamma families; beta and gamma crystallins are also considered as a Calculated MW superfamily. Alpha and beta families are further divided into acidic and basic groups. 20kDa Seven protein regions exist in crystallins: four homologous motifs, a connecting peptide, and N- and C-terminal extensions. Gamma-crystallins are a homogeneous group of Category highly symmetrical, monomeric proteins typically lacking connecting peptides and terminal extensions. They are differentially regulated after early development. Four Primary antibody gamma-crystallin genes (gamma-A through gamma-D) and three pseudogenes (gamma-E, gamma-F, gamma-G) are tandemly organized in a genomic segment as a Applications gene cluster. Whether due to aging or mutations in specific genes, gamma-crystallins WB have been involved in cataract formation. Cross-Reactivity Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 1419 P07316 Immunogen Recombinant fusion protein containing a sequence corresponding to amino acids 80-135 of human CRYGB (NP_005201.2). Synonyms CRYGB;CRYG2;CTRCT39 Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. -

Supplementary Table 3 Complete List of RNA-Sequencing Analysis of Gene Expression Changed by ≥ Tenfold Between Xenograft and Cells Cultured in 10%O2
Supplementary Table 3 Complete list of RNA-Sequencing analysis of gene expression changed by ≥ tenfold between xenograft and cells cultured in 10%O2 Expr Log2 Ratio Symbol Entrez Gene Name (culture/xenograft) -7.182 PGM5 phosphoglucomutase 5 -6.883 GPBAR1 G protein-coupled bile acid receptor 1 -6.683 CPVL carboxypeptidase, vitellogenic like -6.398 MTMR9LP myotubularin related protein 9-like, pseudogene -6.131 SCN7A sodium voltage-gated channel alpha subunit 7 -6.115 POPDC2 popeye domain containing 2 -6.014 LGI1 leucine rich glioma inactivated 1 -5.86 SCN1A sodium voltage-gated channel alpha subunit 1 -5.713 C6 complement C6 -5.365 ANGPTL1 angiopoietin like 1 -5.327 TNN tenascin N -5.228 DHRS2 dehydrogenase/reductase 2 leucine rich repeat and fibronectin type III domain -5.115 LRFN2 containing 2 -5.076 FOXO6 forkhead box O6 -5.035 ETNPPL ethanolamine-phosphate phospho-lyase -4.993 MYO15A myosin XVA -4.972 IGF1 insulin like growth factor 1 -4.956 DLG2 discs large MAGUK scaffold protein 2 -4.86 SCML4 sex comb on midleg like 4 (Drosophila) Src homology 2 domain containing transforming -4.816 SHD protein D -4.764 PLP1 proteolipid protein 1 -4.764 TSPAN32 tetraspanin 32 -4.713 N4BP3 NEDD4 binding protein 3 -4.705 MYOC myocilin -4.646 CLEC3B C-type lectin domain family 3 member B -4.646 C7 complement C7 -4.62 TGM2 transglutaminase 2 -4.562 COL9A1 collagen type IX alpha 1 chain -4.55 SOSTDC1 sclerostin domain containing 1 -4.55 OGN osteoglycin -4.505 DAPL1 death associated protein like 1 -4.491 C10orf105 chromosome 10 open reading frame 105 -4.491 -

Supp Table 1.Pdf
Upregulated genes in Hdac8 null cranial neural crest cells fold change Gene Symbol Gene Title 134.39 Stmn4 stathmin-like 4 46.05 Lhx1 LIM homeobox protein 1 31.45 Lect2 leukocyte cell-derived chemotaxin 2 31.09 Zfp108 zinc finger protein 108 27.74 0710007G10Rik RIKEN cDNA 0710007G10 gene 26.31 1700019O17Rik RIKEN cDNA 1700019O17 gene 25.72 Cyb561 Cytochrome b-561 25.35 Tsc22d1 TSC22 domain family, member 1 25.27 4921513I08Rik RIKEN cDNA 4921513I08 gene 24.58 Ofa oncofetal antigen 24.47 B230112I24Rik RIKEN cDNA B230112I24 gene 23.86 Uty ubiquitously transcribed tetratricopeptide repeat gene, Y chromosome 22.84 D8Ertd268e DNA segment, Chr 8, ERATO Doi 268, expressed 19.78 Dag1 Dystroglycan 1 19.74 Pkn1 protein kinase N1 18.64 Cts8 cathepsin 8 18.23 1500012D20Rik RIKEN cDNA 1500012D20 gene 18.09 Slc43a2 solute carrier family 43, member 2 17.17 Pcm1 Pericentriolar material 1 17.17 Prg2 proteoglycan 2, bone marrow 17.11 LOC671579 hypothetical protein LOC671579 17.11 Slco1a5 solute carrier organic anion transporter family, member 1a5 17.02 Fbxl7 F-box and leucine-rich repeat protein 7 17.02 Kcns2 K+ voltage-gated channel, subfamily S, 2 16.93 AW493845 Expressed sequence AW493845 16.12 1600014K23Rik RIKEN cDNA 1600014K23 gene 15.71 Cst8 cystatin 8 (cystatin-related epididymal spermatogenic) 15.68 4922502D21Rik RIKEN cDNA 4922502D21 gene 15.32 2810011L19Rik RIKEN cDNA 2810011L19 gene 15.08 Btbd9 BTB (POZ) domain containing 9 14.77 Hoxa11os homeo box A11, opposite strand transcript 14.74 Obp1a odorant binding protein Ia 14.72 ORF28 open reading -

A Comprehensive Analysis of the Expression of Crystallins in Mouse Retina Jinghua Xi Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi Washington University School of Medicine in St. Louis Rafal Farjo University of Michigan - Ann Arbor Shigeo Yoshida University of Michigan - Ann Arbor Timothy S. Kern Case Western Reserve University Anand Swaroop University of Michigan - Ann Arbor See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Xi, Jinghua; Farjo, Rafal; Yoshida, Shigeo; Kern, Timothy S.; Swaroop, Anand; and Andley, Usha P., ,"A comprehensive analysis of the expression of crystallins in mouse retina." Molecular Vision.9,. 410-419. (2003). https://digitalcommons.wustl.edu/open_access_pubs/1801 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Jinghua Xi, Rafal Farjo, Shigeo Yoshida, Timothy S. Kern, Anand Swaroop, and Usha P. Andley This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1801 Molecular Vision 2003; 9:410-9 <http://www.molvis.org/molvis/v9/a53> © 2003 Molecular Vision Received 28 May 2003 | Accepted 19 August 2003 | Published 28 August 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi,1 Rafal Farjo,3 Shigeo Yoshida,3 Timothy S. Kern,5 Anand Swaroop,3,4 Usha P. Andley1,2 Departments of 1Ophthalmology and Visual Sciences and 2Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. -

Related Macular Degeneration and Cutis Laxa
UvA-DARE (Digital Academic Repository) Genetic studies of age-related macular degeneration Baas, D.C. Publication date 2012 Document Version Final published version Link to publication Citation for published version (APA): Baas, D. C. (2012). Genetic studies of age-related macular degeneration. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:05 Oct 2021 G������ S������ �� A��-������� M������ D����������� D����������� M������ G������ S������ �� A��-������� | 2012 D�������� C. B��� G������ S������ �� A��-������� M������ D����������� D�������� C. B��� cover.indd 1 31-10-12 08:36 Genetic Studies of Age-related Macular Degeneration Dominique C. Baas Chapter 0.indd 1 23-10-12 19:24 The research described in this thesis was conducted at the Netherlands Institute for Neuroscience (NIN), an institute of the Royal Netherlands Academy of Arts and Sciences, Department of Clinical and Molecular Ophthalmogenetics, Amsterdam, The Netherlands. -

Congenital Cataracts Due to a Novel 2‑Bp Deletion in CRYBA1/A3
1614 MOLECULAR MEDICINE REPORTS 10: 1614-1618, 2014 Congenital cataracts due to a novel 2‑bp deletion in CRYBA1/A3 JING ZHANG1, YANHUA ZHANG1, FANG FANG1, WEIHONG MU1, NING ZHANG2, TONGSHUN XU3 and QINYING CAO1 1Prenatal Diagnosis Center, Shijiazhuang Obstetrics and Gynecology Hospital; 2Department of Cardiology, The Second Hospital of Hebei Medical University; 3Department of Surgery, Shijiazhuang Obstetrics and Gynecology Hospital, Shijiazhuang, Hebei, P.R. China Received September 22, 2013; Accepted April 11, 2014 DOI: 10.3892/mmr.2014.2324 Abstract. Congenital cataracts, which are a clinically and located in the eye lens. The major human crystallins comprise genetically heterogeneous group of eye disorders, lead to 90% of protein in the mature lens and contain two different visual impairment and are a significant cause of blindness superfamilies: the small heat‑shock proteins (α-crystallins) in childhood. A major proportion of the causative mutations and the βγ-crystallins. for congenital cataracts are found in crystallin genes. In the In this study a functional candidate approach was used present study, a novel deletion mutation (c.590-591delAG) in to investigate the known crystallin genes, including CRYAA, exon 6 of CRYBA1/A3 was identified in a large family with CRYAB, CRYBA1/A3, CRYBB1, CRYBB2, CRYGC, CRYGD autosomal dominant congenital cataracts. An increase in and CRYGS, in which a major proportion of the mutations local hydrophobicity was predicted around the mutation site; identified in a large family with congenital cataracts were however, further studies are required to determine the exact found. effect of the mutation on βA1/A3-crystallin structure and function. To the best of our knowledge, this is the first report Subjects and methods of an association between a frameshift mutation in exon 6 of CRYBA1/A3 and congenital cataracts. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

Congenital Eye Disorders Gene Panel
Congenital eye disorders gene panel Contact details Introduction Regional Genetics Service Ocular conditions are highly heterogeneous and show considerable phenotypic overlap. 1 in Levels 4-6, Barclay House 2,500 children in the UK are diagnosed as blind or severely visually impaired by the time they 37 Queen Square reach one year old. As many as half of these cases are likely to be inherited and remain undiagnosed due to the vast number of genes involved in these conditions. Many congenital London, WC1N 3BH eye disorders causing visual impairment or blindness at birth or progressive visual impairment T +44 (0) 20 7762 6888 also include syndromic conditions involving additional metabolic, developmental, physical or F +44 (0) 20 7813 8578 sensory abnormalities. Gene panels offer the enhanced probability of diagnosis as a very large number of genes can be interrogated. Samples required Ocular birth defects include all inheritance modalities. Autosomal dominant and recessive 5ml venous blood in plastic EDTA diseases as well as X-linked dominant and recessive diseases are seen. These conditions can bottles (>1ml from neonates) also be caused by de novo variants. Prenatal testing must be arranged Referrals in advance, through a Clinical Genetics department if possible. Patients presenting with a phenotype appropriate for the requested sub-panel Amniotic fluid or CV samples Referrals will be accepted from clinical geneticists and consultants in ophthalmology. should be sent to Cytogenetics for Prenatal testing dissecting and culturing, with instructions to forward the sample Prenatal diagnosis may be offered as appropriate where pathogenic variants have been to the Regional Molecular Genetics identified in accordance with expected inheritance pattern and where appropriate parental laboratory for analysis testing and counselling has been conducted. -

Duplication 9P and Their Implication to Phenotype
Guilherme et al. BMC Medical Genetics (2014) 15:142 DOI 10.1186/s12881-014-0142-1 RESEARCH ARTICLE Open Access Duplication 9p and their implication to phenotype Roberta Santos Guilherme1, Vera Ayres Meloni1, Ana Beatriz Alvarez Perez1, Ana Luiza Pilla1, Marco Antonio Paula de Ramos1, Anelisa Gollo Dantas1, Sylvia Satomi Takeno1, Leslie Domenici Kulikowski2 and Maria Isabel Melaragno1* Abstract Background: Trisomy 9p is one of the most common partial trisomies found in newborns. We report the clinical features and cytogenomic findings in five patients with different chromosome rearrangements resulting in complete 9p duplication, three of them involving 9p centromere alterations. Methods: The rearrangements in the patients were characterized by G-banding, SNP-array and fluorescent in situ hybridization (FISH) with different probes. Results: Two patients presented de novo dicentric chromosomes: der(9;15)t(9;15)(p11.2;p13) and der(9;21)t(9;21) (p13.1;p13.1). One patient presented two concomitant rearranged chromosomes: a der(12)t(9;12)(q21.13;p13.33) and an psu i(9)(p10) which showed FISH centromeric signal smaller than in the normal chromosome 9. Besides the duplication 9p24.3p13.1, array revealed a 7.3 Mb deletion in 9q13q21.13 in this patient. The break in the psu i(9) (p10) probably occurred in the centromere resulting in a smaller centromere and with part of the 9q translocated to the distal 12p with the deletion 9q occurring during this rearrangement. Two patients, brother and sister, present 9p duplication concomitant to 18p deletion due to an inherited der(18)t(9;18)(p11.2;p11.31)mat. -

Transgenic Zebrafish Models Reveal Distinct Molecular Mechanisms for Cataract-Linked Αa-Crystallin Mutants
bioRxiv preprint doi: https://doi.org/10.1101/364125; this version posted July 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Transgenic Zebrafish Models Reveal Distinct Molecular Mechanisms for Cataract-linked αA-Crystallin Mutants Shu-Yu Wu, Ping Zou, Sanjay Mishra, Hassane S Mchaourab* Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 37232, USA Running title: Distinct mechanisms of α-crystallin mutations * Corresponding author: Hassane Mchaourab 741 Light Hall 2215 Garland Avenue Molecular Physiology & Biophysics Nashville, TN 37232 Office: 615.322.3307 Fax: 615.322.7236 Email: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/364125; this version posted July 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Mutations in the small heat shock proteins a-crystallins have been linked to autosomal dominant cataracts in humans. Extensive studies in vitro have revealed a spectrum of alterations to the structure and function of these proteins including shifts in the size of the oligomer, modulation of subunit exchange and modification of their affinity to client proteins. Although mouse models of these mutants were instrumental in identifying changes in cellular proliferation and lens development, a direct comparative analysis of their effects on lens proteostasis has not been performed. -

Supplementary Materials
Supplementary materials Gene Nucleotide Amino acid change Clinical phenotype Ref symbol change CRYAA c.35G>T p. R12L lens protein gene [1] CRYAB c.32G>A p. R11H lens protein gene [2] CRYBA1 c.279-281delG p.ΔG91 lens protein gene [3] AG CRYBA4 c.206T>C p. L69P lens protein gene [4] CRYBB1 c.658G>T p. G220X lens protein gene [5] CRYBB2 c.563G>A p. R188H lens protein gene [6] CRYBB3 c.314G>A p. R105Q lens protein gene [7] CRYGA c.196T>C p. Y66H lens protein gene [8] CRYGB c.449G>T p. G150V lens protein gene [8] CRYGC c.385G>T p. G129C lens protein gene [9] CRYGD c.70C>A p. P24T lens protein gene [10] CRYGS c.53G>T :p.G18V lens protein gene [11] GJA3 c.188A>G p.N63S membrane protein [12] gene GJA8 c.262C>T p.P88S membrane protein [13] gene BFSP1 c736-1384_c.9 T246fsX7 cytoskeleton protein [14] 57-66del gene BFSP2 c.1091G>A p.G364D cytoskeleton protein [15] gene PAX6 c.307C>T p.R103X developmental [16] regulatory protein gen PITX3 c.38G>A p.S13N developmental [17] regulatory protein gen HSF4 c.524G>C p.R175P developmental [18] regulatory protein gen MAF c.863G>C p.R288P developmental [19] regulatory protein gen CHMP4B c.481G>A p. E161K chromatin modified [20] protein gene EPHA2 c.2842G>T p. G948W tyrosine kinase [21] receptor gene COL4A1 c.2345G>C p. G782A syndrome-related [22] genes FTL c.160G>A p.E54K developmental [23] regulatory protein gen GALK1 c.416T>C p.