A Large Effective Population Size for Established Within-Host Influenza Virus Infection Casper K Lumby1, Lei Zhao1, Judith Breuer2,3, Christopher JR Illingworth1,4,5*
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Mutational Load Causes Stochastic Evolutionary Outcomes In
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Apollo Virus Evolution, 2019, 5(1): vez008 doi: 10.1093/ve/vez008 Research article Mutational load causes stochastic evolutionary outcomes in acute RNA viral infection Downloaded from https://academic.oup.com/ve/article-abstract/5/1/vez008/5476199 by guest on 01 June 2020 Lei Zhao,1,† Ali B. Abbasi,1 and Christopher J. R. Illingworth1,2,*,‡ 1Department of Genetics, University of Cambridge, Cambridge, UK and 2Department of Applied Mathematics and Theoretical Physics, University of Cambridge, Cambridge, UK *Corresponding author: E-mail: [email protected] †http://orcid.org/0000-0002-6551-2707 ‡http://orcid.org/0000-0002-0030-2784 Abstract Mutational load is known to be of importance for the evolution of RNA viruses, the combination of a high mutation rate and large population size leading to an accumulation of deleterious mutations. However, while the effects of mutational load on global viral populations have been considered, its quantitative effects at the within-host scale of infection are less well un- derstood. We here show that even on the rapid timescale of acute disease, mutational load has an effect on within-host vi- ral adaptation, reducing the effective selection acting upon beneficial variants by 10 per cent. Furthermore, mutational load induces considerable stochasticity in the pattern of evolution, causing a more than five-fold uncertainty in the effective fitness of a transmitted beneficial variant. Our work aims to bridge the gap between classic models from population genetic theory and the biology of viral infection. -

G2 Phase Cell Cycle Regulation by E2F4 Following Genotoxic Stress
G2 Phase Cell Cycle Regulation by E2F4 Following Genotoxic Stress by MEREDITH ELLEN CROSBY Submitted in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Thesis Advisor: Dr. Alex Almasan Department of Environmental Health Sciences CASE WESTERN RESERVE UNIVERSITY May, 2006 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the dissertation of ______________________________________________________ candidate for the Ph.D. degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. TABLE OF CONTENTS TABLE OF CONTENTS………………………………………………………………….1 LIST OF FIGURES……………………………………………………………………….5 LIST OF TABLES………………………………………………………………………...7 ACKNOWLEDGEMENTS……………………………………………………………….8 LIST OF ABBREVIATIONS……………………………………………………………10 ABSTRACT……………………………………………………………………………...15 CHAPTER 1. INTRODUCTION 1.1. CELL CYCLE REGULATION: HISTORICAL OVERVIEW………………...17 1.2. THE E2F FAMILY OF TRANSCRIPTION FACTORS……………………….22 1.3. E2F AND CELL CYCLE CONTROL 1.3.1. G0/G1 Phase Transition………………………………………………….28 1.3.2. S Phase…………………………………………………………………...28 1.3.3. G2/M Phase Transition…………………………………………………..30 1.4. -

Population Size and the Rate of Evolution
Review Population size and the rate of evolution 1,2 1 3 Robert Lanfear , Hanna Kokko , and Adam Eyre-Walker 1 Ecology Evolution and Genetics, Research School of Biology, Australian National University, Canberra, ACT, Australia 2 National Evolutionary Synthesis Center, Durham, NC, USA 3 School of Life Sciences, University of Sussex, Brighton, UK Does evolution proceed faster in larger or smaller popu- mutations occur and the chance that each mutation lations? The relationship between effective population spreads to fixation. size (Ne) and the rate of evolution has consequences for The purpose of this review is to synthesize theoretical our ability to understand and interpret genomic varia- and empirical knowledge of the relationship between tion, and is central to many aspects of evolution and effective population size (Ne, Box 1) and the substitution ecology. Many factors affect the relationship between Ne rate, which we term the Ne–rate relationship (NeRR). A and the rate of evolution, and recent theoretical and positive NeRR implies faster evolution in larger popula- empirical studies have shown some surprising and tions relative to smaller ones, and a negative NeRR implies sometimes counterintuitive results. Some mechanisms the opposite (Figure 1A,B). Although Ne has long been tend to make the relationship positive, others negative, known to be one of the most important factors determining and they can act simultaneously. The relationship also the substitution rate [5–8], several novel predictions and depends on whether one is interested in the rate of observations have emerged in recent years, causing some neutral, adaptive, or deleterious evolution. Here, we reassessment of earlier theory and highlighting some gaps synthesize theoretical and empirical approaches to un- in our understanding. -

Individual Index
INDIVIDUAL INDEX Bl Page Page A Cooremans-Cruz, Eugenio Juan 3825 Cortes, Eugenia 2474 Abbott, Charles P 3807 Crowley, Daniel 3820 Abrams, Samuel S 3825 Cruz, Marinelle Khristy 3836 Abshire, Arthur J 3812 Cuervo, Ester 3825 Ah Kwang Chang 3827 Cuffy, Jackson Ormiston Edwards and Ah Sing Ying 3826 Merle Cleopatra Edwards 38J54 Ah Young Cho Kwak 3803 Alcala-Salcedo, Apolinario 3825 D Alvarado, Irma Victoria Bolarte 3853 Araujo,Lilia 3828 D'Souza, Eustace John 3824 Araujo, Maria Freitas 3825 Datronics Engineers, Inc 3857 Arellano, Santos Marquez 3830 Daves, Gary 3862 Armstrong, Anne L 235 Davies, Mrs. Olive M.V.T., Samira Arriola, Rodolfo N 3851 D.K., Ola-Tomi K., Ola-Yinka K., Asencio-Placencio, Pedro 3825 Ilesha E.K., and Baba-Tunji K 3803 Atherley, Juana Todd 3829 Davis, Jefferson F 1304 Austin, John P 234 Day, Craig 3824 DeBravo, Cenovia Mesa 3825 B DeLuna-Segovia, Jose 3825 Dibben, James William 3823 Bader, Louis William 3825 Din, Ruben P 3831 Baglieri, George 3825 Do Sook Park 3841 Bakierowska, Kazimiera 3827 Dom Min Lee 3826 Bally, George Ally 3827 Dowd, Kazuko Nishioka 3820 Barengo, Bernardino 2485 Barrera-Cabrera, Jesus 3825 Batavia Turf Farms, Inc 3818 • ^- • E Benney, Donna Marainne 3836 Eaker, Lt. Gen. IraC 1060 Berger, Harry 3825 Eha,Elmar 3825 Binabise, Juanita 3840 Ehard,Karin 3858 Birely, Victor M 1643 Elder, William J 3814 Blakeley, Hildegard G 3845 Emde, Noel Abueg 3837 Blanquicett, Carmen Cecilia 3859 Encomienda, Rogelio M 3853 Bonderenko, Edward J 2485 Espenueva, Edmundo Alfredo Boone, Master Sgt. William E 3813 Oreiro -

Transformations of Lamarckism Vienna Series in Theoretical Biology Gerd B
Transformations of Lamarckism Vienna Series in Theoretical Biology Gerd B. M ü ller, G ü nter P. Wagner, and Werner Callebaut, editors The Evolution of Cognition , edited by Cecilia Heyes and Ludwig Huber, 2000 Origination of Organismal Form: Beyond the Gene in Development and Evolutionary Biology , edited by Gerd B. M ü ller and Stuart A. Newman, 2003 Environment, Development, and Evolution: Toward a Synthesis , edited by Brian K. Hall, Roy D. Pearson, and Gerd B. M ü ller, 2004 Evolution of Communication Systems: A Comparative Approach , edited by D. Kimbrough Oller and Ulrike Griebel, 2004 Modularity: Understanding the Development and Evolution of Natural Complex Systems , edited by Werner Callebaut and Diego Rasskin-Gutman, 2005 Compositional Evolution: The Impact of Sex, Symbiosis, and Modularity on the Gradualist Framework of Evolution , by Richard A. Watson, 2006 Biological Emergences: Evolution by Natural Experiment , by Robert G. B. Reid, 2007 Modeling Biology: Structure, Behaviors, Evolution , edited by Manfred D. Laubichler and Gerd B. M ü ller, 2007 Evolution of Communicative Flexibility: Complexity, Creativity, and Adaptability in Human and Animal Communication , edited by Kimbrough D. Oller and Ulrike Griebel, 2008 Functions in Biological and Artifi cial Worlds: Comparative Philosophical Perspectives , edited by Ulrich Krohs and Peter Kroes, 2009 Cognitive Biology: Evolutionary and Developmental Perspectives on Mind, Brain, and Behavior , edited by Luca Tommasi, Mary A. Peterson, and Lynn Nadel, 2009 Innovation in Cultural Systems: Contributions from Evolutionary Anthropology , edited by Michael J. O ’ Brien and Stephen J. Shennan, 2010 The Major Transitions in Evolution Revisited , edited by Brett Calcott and Kim Sterelny, 2011 Transformations of Lamarckism: From Subtle Fluids to Molecular Biology , edited by Snait B. -

Changes in Population Dynamics in Mutualistic Versus Pathogenic Viruses
Viruses 2011, 3, 12-19; doi:10.3390/v3010012 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Commentary Changes in Population Dynamics in Mutualistic versus Pathogenic Viruses Marilyn J. Roossinck Plant Biology Division, The Samuel Roberts Noble Foundation, Inc., P.O. Box 2180, Ardmore, OK 73402, USA; E-Mail: [email protected]; Tel.: +1 580 224 6600; Fax: +1 580 224 6692. Received: 17 December 2010; in revised form: 31 December 2010 / Accepted: 6 January 2011 / Published: 17 January 2011 Abstract: Although generally regarded as pathogens, viruses can also be mutualists. A number of examples of extreme mutualism (i.e., symbiogenesis) have been well studied. Other examples of mutualism are less common, but this is likely because viruses have rarely been thought of as having any beneficial effects on their hosts. The effect of mutualism on the population dynamics of viruses is a topic that has not been addressed experimentally. However, the potential for understanding mutualism and how a virus might become a mutualist may be elucidated by understanding these dynamics. Keywords: beneficial viruses; polymerase fidelity; quasispecies; symbiosis; symbiogenesis 1. Introduction Viruses have been studied predominantly as pathogens, beginning with the first virus ever described, Tobacco mosaic virus [1] that was causing spots on tobacco plants. However, a number of viruses in plants, animals, fungi and bacteria have been described that are not pathogens; many are commensals and some are mutualists. Traditionally, mutualistic symbioses are thought of as long-term stable relationships, but viruses can clearly switch lifestyles depending on conditions. What effect does mutualism have on the population dynamics of a virus? Do the population dynamics of conditional mutualists change depending on their lifestyle? This is the subject of this brief perspective. -

IN EVOLUTION JACK LESTER KING UNIVERSITY of CALIFORNIA, SANTA BARBARA This Paper Is Dedicated to Retiring University of California Professors Curt Stern and Everett R
THE ROLE OF MUTATION IN EVOLUTION JACK LESTER KING UNIVERSITY OF CALIFORNIA, SANTA BARBARA This paper is dedicated to retiring University of California Professors Curt Stern and Everett R. Dempster. 1. Introduction Eleven decades of thought and work by Darwinian and neo-Darwinian scientists have produced a sophisticated and detailed structure of evolutionary ,theory and observations. In recent years, new techniques in molecular biology have led to new observations that appear to challenge some of the basic theorems of classical evolutionary theory, precipitating the current crisis in evolutionary thought. Building on morphological and paleontological observations, genetic experimentation, logical arguments, and upon mathematical models requiring simplifying assumptions, neo-Darwinian theorists have been able to make some remarkable predictions, some of which, unfortunately, have proven to be inaccurate. Well-known examples are the prediction that most genes in natural populations must be monomorphic [34], and the calculation that a species could evolve at a maximum rate of the order of one allele substitution per 300 genera- tions [13]. It is now known that a large proportion of gene loci are polymorphic in most species [28], and that evolutionary genetic substitutions occur in the human line, for instance, at a rate of about 50 nucleotide changes per generation [20], [24], [25], [26]. The puzzling observation [21], [40], [46], that homologous proteins in different species evolve at nearly constant rates is very difficult to account for with classical evolutionary theory, and at the very least gives a solid indication that there are qualitative differences between the ways molecules evolve and the ways morphological structures evolve. -
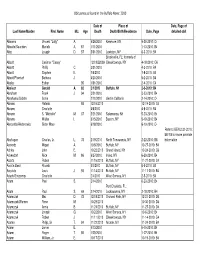
Obituaries Buffalo News 2010 by Name
Obituaries as found in the Buffalo News: 2010 Date of Place of Date, Page of Last Name/Maiden First Name M.I. Age Death Death/Birth/Residence Date, Page detailed obit Abbarno Vincent "Lolly" A. 9/26/2010 Kenmore, NY 9-30-2010: C4 Abbatte/Saunders Murielle A. 87 1/11/2010 1-13-2010: B4 Abbo Joseph D. 57 5/31/2010 Lewiston, NY 6-3-2010: B4 Brooksville, FL; formerly of Abbott Casimer "Casey" 12/19/22009 Cheektowaga, NY 4-18-2010: C6 Abbott Phillip C. 3/31/2010 4-3-2010: B4 Abbott Stephen E. 7/6/2010 7-8-2010: B4 Abbott/Pfoetsch Barbara J. 4/20/2010 5-2-2010: B4 Abeles Esther 95 1/31/2010 2-4-2010: C4 Abelson Gerald A. 82 2/1/2010 Buffalo, NY 2-3-2010: B4 Abraham Frank J. 94 3/21/2010 3-23-2010: B4 Abrahams/Gichtin Sonia 2/10/2010 died in California 2-14-2010: C4 Abramo Rafeala 93 12/16/2010 12-19-2010: C4 Abrams Charlotte 4/6/2010 4-8-2010: B4 Abrams S. "Michelle" M. 37 5/21/2010 Salamanca, NY 5-23-2010: B4 Abrams Walter I. 5/15/2010 Basom, NY 5-19-2010: B4 Abrosette/Aksterowicz Sister Mary 6/18/2010 6-19-2010: C4 Refer to BEN 2-21-2010: B6/7/8 for more possible Abshagen Charles, Jr. L. 73 2/19/2010 North Tonawanda, NY 2-22-2010: B8 information Acevedo Miguel A. 10/6/2010 Buffalo, NY 10-27-2010: B4 Achkar John E. -

Men Final Entries
Final Entries - Athletes List by event European Athletics Indoor Championships 2021, Torun/POL Tot. Number of countries Tot. Number of athletes Tot. Number of Men Tot. Number of Women 47 733 405 328 FINAL ENTRIES - Men 60m Senior Men Num. of countries: 33 Num. of athletes: 71 Member Federation Surname First Name DoB PB SB ARM Donigian Alexander 20/10/1993 6.64i 6.79i ART Keletela Dorian Celeste 06/02/1999 6.79i 6.85i AUT Fuchs Markus 14/11/1995 6.62i 6.69i BEL Kuba Di-Vita Gaylord 17/11/1995 6.73i 6.75i BEL Vleminckx Kobe 31/05/1998 6.65i 6.65i BLR Bliznets Dzianis 12/03/1995 6.75i 6.75i BLR Bohdan Maksim 19/03/1997 6.77i 6.77i BLR Zabalotny Yury 24/02/1997 6.72i 6.72i BUL Dimitrov Denis 10/02/1994 6.65i 6.73i BUL Jivkov Vesselin 26/01/2001 6.76i 6.80i CZE Hampl Štěpán 10/11/1999 6.70i 6.70i CZE Stromšík Zdeněk 25/11/1994 6.60i 6.68i CZE Veleba Jan 06/12/1986 6.65i 6.65i DEN Hansen Simon 30/06/1998 6.75i 6.75i DEN Kjær Emil Mader 20/12/1999 6.77i 6.77i DEN Musah Kojo 15/04/1996 6.61i 6.61i ESP López Sergio 05/07/1999 6.67i 6.74i ESP Rodríguez Daniel 26/01/1995 6.67i 6.67i ESP Sanchez Ricardo 10/08/1999 6.75i 6.75i EST Nazarov Karl Erik 17/03/1999 6.63i 6.63i FIN Illukka Riku 21/09/1999 6.73i 6.73i FIN Purola Samuel 19/05/2000 6.67i 6.67i FIN Samuelsson Samuli 23/06/1995 6.66i 6.66i FRA Fall Mouhamadou 25/02/1992 6.62i 6.62i FRA Golitin Amaury 28/01/1997 6.62i 6.62i GBR Aikines-Aryeetey Harry 29/08/1988 6.55i 6.67i GBR Bromby Oliver 30/03/1998 6.63i 6.65i GBR Robertson Andrew 17/12/1990 6.54i 6.61i GER Corucle Philipp 18/07/1997 6.62i -

Evolutionary Analysis of the Dynamics of Viral Infectious Disease
REVIEWS MODELLING Evolutionary analysis of the dynamics of viral infectious disease Oliver G. Pybus* and Andrew Rambaut‡ Abstract | Many organisms that cause infectious diseases, particularly RNA viruses, mutate so rapidly that their evolutionary and ecological behaviours are inextricably linked. Consequently, aspects of the transmission and epidemiology of these pathogens are imprinted on the genetic diversity of their genomes. Large-scale empirical analyses of the evolutionary dynamics of important pathogens are now feasible owing to the increasing availability of pathogen sequence data and the development of new computational and statistical methods of analysis. In this Review, we outline the questions that can be answered using viral evolutionary analysis across a wide range of biological scales. REFS 5–7 Balancing selection Rapidly evolving pathogens are unique in that their key human pathogens (for example, ). Any form of natural selection ecological and evolutionary dynamics occur on the Understandably, most studies have focused on impor- that results in the maintenance same timescale and can therefore potentially interact. tant human RNA viruses such as influenza virus, HIV, of genetic polymorphisms in a For example, the exceptionally high nucleotide mutation dengue virus and hepatitis C virus (HCV); therefore, this population, as opposed to 1 their loss through fixation or rate of a typical RNA virus — a million times greater Review concentrates on these infections. However, the elimination. than that of vertebrates — allows these viruses to gener- range of pathogens and hosts to which phylodynamic ate mutations and adaptations de novo during environ- methods are applied is expanding, and we also discuss mental change, whereas other organisms must rely on infectious diseases of wildlife, crops and livestock. -

Sales Report
Sales Listing Report Page 1 of 262 McHenry County 01/21/2021 08:25:40 01/01/2020 - 09/30/2020 Township: GRAFTON TWP Document Number Sale Year Sale Type Valid Sale Sale Date Dept. Study Selling Price Parcel Number Built Year Property Type Prop. Class Acres Square Ft. Lot Size Grantor Name Grantee Name Site Address 2020R0006895 2020 Warranty Deed Y 02/13/2020 Y $189,000.00 REINVEST HOMES, LLC ANGELA STROPE 160 S HEATHER DR 18-01-101-027 0 GARAGE/ NO O O 0040 .00 0 CRYSTAL LAKE, IL 600145177 160 S HEATHER DR CRYSTAL LAKE, IL 60014 - Legal Description: DOC 2020R0006895 LT 31 BLK 15 R A CEPEKS CRYSTAL VISTA 2020R0030830 2020 Partial Assessmen N 07/30/2020 N $136,500.00 DANIEL M ADAMS, AS TRUST BRETT A RIDINGS 1324 THORNWOOD LN 18-01-102-037 0 GARAGE/ NO O O 0040 .00 0 CRYSTAL LAKE, IL 600145042 TARI D RIDINGS 1324 THORNWOOD LANE CRYSTAL LAKE, IL 600140000 1324 THORNWOOD LN CRYSTAL LAKE, IL 60014 -5042 Legal Description: DOC 2020R0030830 LT 1 & PT LT 19 LYING N OF & ADJ BLK 14 R A CEPEKS CRYSTAL VISTA 2020R0010709 2020 Warranty Deed Y 03/16/2020 Y $210,000.00 PATRICK J. CARAMELA JUAN P. MIRANDA STACY C. CARAMELA 58 HOLLY DR 18-01-106-018 0 LDG SINGLE FAM 0040 .00 0 CRYSTAL LAKE, IL 600145022 58 HOLLY DR CRYSTAL LAKE, IL 60014 -5022 Legal Description: DOC 2020R0010709 LT 7 BLK 19 R A CEPEKS CRYSTAL VISTA 2020R0015717 2020 Warranty Deed Y 04/23/2020 Y $95,000.00 LISA M SULMA FANO THEOFANOUS 5011 NORTHWEST HWY 18-01-107-011 0 LDG SINGLE FAM 0040 .00 0 CRYSTAL LAKE, IL 600147330 STEVE THEOFANOUS 5011 NORTHWEST HIGHWAY CRYSTAL LAKE, IL 600147330 113 HEATHER DR CRYSTAL LAKE, IL 60014 - Legal Description: DOC 2020R0015717 LT 12 BLK 31 R A CEPEKS CRYSTAL VISTA Copyright (C) 1997-2021 DEVNET Incorporated MXSTIEG Sales Listing Report Page 2 of 262 McHenry County 01/21/2021 08:25:40 01/01/2020 - 09/30/2020 Township: GRAFTON TWP Document Number Sale Year Sale Type Valid Sale Sale Date Dept. -
![Arxiv:1810.00224V2 [Q-Bio.PE] 7 Dec 2020 Humanity Is Increasingly Influencing Global Environments [195]](https://docslib.b-cdn.net/cover/3556/arxiv-1810-00224v2-q-bio-pe-7-dec-2020-humanity-is-increasingly-in-uencing-global-environments-195-943556.webp)
Arxiv:1810.00224V2 [Q-Bio.PE] 7 Dec 2020 Humanity Is Increasingly Influencing Global Environments [195]
A Survey of Biodiversity Informatics: Concepts, Practices, and Challenges Luiz M. R. Gadelha Jr.1* Pedro C. de Siracusa1 Artur Ziviani1 Eduardo Couto Dalcin2 Helen Michelle Affe2 Marinez Ferreira de Siqueira2 Luís Alexandre Estevão da Silva2 Douglas A. Augusto3 Eduardo Krempser3 Marcia Chame3 Raquel Lopes Costa4 Pedro Milet Meirelles5 and Fabiano Thompson6 1National Laboratory for Scientific Computing, Petrópolis, Brazil 2Friedrich-Schiller-University Jena, Jena, Germany 2Rio de Janeiro Botanical Garden, Rio de Janeiro, Brazil 3Oswaldo Cruz Foundation, Rio de Janeiro, Brazil 4National Institute of Cancer, Rio de Janeiro, Brazil 5Federal University of Bahia, Salvador, Brazil 6Federal University of Rio de Janeiro, Rio de Janeiro, Brazil Abstract The unprecedented size of the human population, along with its associated economic activities, have an ever increasing impact on global environments. Across the world, countries are concerned about the growing resource consumption and the capacity of ecosystems to provide them. To effectively conserve biodiversity, it is essential to make indicators and knowledge openly available to decision-makers in ways that they can effectively use them. The development and deployment of mechanisms to produce these indicators depend on having access to trustworthy data from field surveys and automated sensors, biological collections, molec- ular data, and historic academic literature. The transformation of this raw data into synthesized information that is fit for use requires going through many refinement steps. The methodologies and techniques used to manage and analyze this data comprise an area often called biodiversity informatics (or e-Biodiversity). Bio- diversity data follows a life cycle consisting of planning, collection, certification, description, preservation, discovery, integration, and analysis.