Novel Bicyclic and Tricyclic Cannabinergic Ligands As
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Modifications to the Harmonized Tariff Schedule of the United States to Implement Changes to the Pharmaceutical Appendix
United States International Trade Commission Modifications to the Harmonized Tariff Schedule of the United States to Implement Changes to the Pharmaceutical Appendix USITC Publication 4208 December 2010 U.S. International Trade Commission COMMISSIONERS Deanna Tanner Okun, Chairman Irving A. Williamson, Vice Chairman Charlotte R. Lane Daniel R. Pearson Shara L. Aranoff Dean A. Pinkert Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Modifications to the Harmonized Tariff Schedule of the United States to Implement Changes to the Pharmaceutical Appendix Publication 4208 December 2010 (This page is intentionally blank) Pursuant to the letter of request from the United States Trade Representative of December 15, 2010, set forth at the end of this publication, and pursuant to section 1207(a) of the Omnibus Trade and Competitiveness Act, the United States International Trade Commission is publishing the following modifications to the Harmonized Tariff Schedule of the United States (HTS) to implement changes to the Pharmaceutical Appendix, effective on January 1, 2011. Table 1 International Nonproprietary Name (INN) products proposed for addition to the Pharmaceutical Appendix to the Harmonized Tariff Schedule INN CAS Number Abagovomab 792921-10-9 Aclidinium Bromide 320345-99-1 Aderbasib 791828-58-5 Adipiplon 840486-93-3 Adoprazine 222551-17-9 Afimoxifene 68392-35-8 Aflibercept 862111-32-8 Agatolimod -

Mitochondrial ADP/ATP Exchange Inhibition
www.nature.com/scientificreports OPEN Mitochondrial ADP/ATP exchange inhibition: a novel off-target mechanism underlying ibipinabant- Received: 27 March 2015 Accepted: 27 August 2015 induced myotoxicity Published: 29 September 2015 Tom J. J. Schirris1,2, Tina Ritschel3, G. Herma Renkema2,4, Peter H. G. M. Willems2,5, Jan A. M. Smeitink2,4 & Frans G. M. Russel1,2 Cannabinoid receptor 1 (CB1R) antagonists appear to be promising drugs for the treatment of obesity, however, serious side effects have hampered their clinical application. Rimonabant, the first in class CB1R antagonist, was withdrawn from the market because of psychiatric side effects. This has led to the search for more peripherally restricted CB1R antagonists, one of which is ibipinabant. However, this 3,4-diarylpyrazoline derivative showed muscle toxicity in a pre-clinical dog study with mitochondrial dysfunction. Here, we studied the molecular mechanism by which ibipinabant induces mitochondrial toxicity. We observed a strong cytotoxic potency of ibipinabant in C2C12 myoblasts. Functional characterization of mitochondria revealed increased cellular reactive oxygen species generation and a decreased ATP production capacity, without effects on the catalytic activities of mitochondrial enzyme complexes I–V or the complex specific-driven oxygen consumption. Using in silico off-target prediction modelling, combined with in vitro validation in isolated mitochondria and mitoplasts, we identified adenine nucleotide translocase (ANT)-dependent mitochondrial ADP/ATP exchange as a novel molecular mechanism underlying ibipinabant-induced toxicity. Minor structural modification of ibipinabant could abolish ANT inhibition leading to a decreased cytotoxic potency, as observed with the ibipinabant derivative CB23. Our results will be instrumental in the development of new types of safer CB1R antagonists. -

Potential Cannabis Antagonists for Marijuana Intoxication
Central Journal of Pharmacology & Clinical Toxicology Bringing Excellence in Open Access Review Article *Corresponding author Matthew Kagan, M.D., Cedars-Sinai Medical Center, 8730 Alden Drive, Los Angeles, CA 90048, USA, Tel: 310- Potential Cannabis Antagonists 423-3465; Fax: 310.423.8397; Email: Matthew.Kagan@ cshs.org Submitted: 11 October 2018 for Marijuana Intoxication Accepted: 23 October 2018 William W. Ishak, Jonathan Dang, Steven Clevenger, Shaina Published: 25 October 2018 Ganjian, Samantha Cohen, and Matthew Kagan* ISSN: 2333-7079 Cedars-Sinai Medical Center, USA Copyright © 2018 Kagan et al. Abstract OPEN ACCESS Keywords Cannabis use is on the rise leading to the need to address the medical, psychosocial, • Cannabis and economic effects of cannabis intoxication. While effective agents have not yet been • Cannabinoids implemented for the treatment of acute marijuana intoxication, a number of compounds • Antagonist continue to hold promise for treatment of cannabinoid intoxication. Potential therapeutic • Marijuana agents are reviewed with advantages and side effects. Three agents appear to merit • Intoxication further inquiry; most notably Cannabidiol with some evidence of antipsychotic activity • THC and in addition Virodhamine and Tetrahydrocannabivarin with a similar mixed receptor profile. Given the results of this research, continued development of agents acting on cannabinoid receptors with and without peripheral selectivity may lead to an effective treatment for acute cannabinoid intoxication. Much work still remains to develop strategies that will interrupt and reverse the effects of acute marijuana intoxication. ABBREVIATIONS Therapeutic uses of cannabis include chronic pain, loss of appetite, spasticity, and chemotherapy-associated nausea and CBD: Cannabidiol; CBG: Cannabigerol; THCV: vomiting [8]. Recreational cannabis use is on the rise with more Tetrahydrocannabivarin; THC: Tetrahydrocannabinol states approving its use and it is viewed as no different from INTRODUCTION recreational use of alcohol or tobacco [9]. -

Potential Health Benefits of Cannabis Extracts: a Review Maria Rosana Ramirez
International Journal of Chemical and Biomedical Science 2016; 2(1): 1-8 Published online March 1, 2016 (http://www.aascit.org/journal/ijcbs) Potential Health Benefits of Cannabis Extracts: A Review Maria Rosana Ramirez CITER-The National Scientific and Technical Research Council (CONICET), Argentine Government, Agency, Argentina Email address [email protected] Citation Maria Rosana Ramirez. Potential Health Benefits of Cannabis Extracts: A Review. International Journal of Chemical and Biomedical Science . Vol. 2, No. 1, 2016, pp. 1-8. Keywords Abstract Non-cannabinoids Compounds, A central tenet underlying the use of plant preparations is that herbs contain many Cannabis Extracts, bioactive compounds. Cannabis contains tetrahydrocannabinols (THC) a primary Bioactivity metabolite with reported psychotropic effects. Therefore, the presence of THC makes controversial the use of Cannabis to treat diseases by which their uses and applications were limited. The question then is: is it possible to use the extracts from Cannabis to treat the diseases related with it use in folk medicine? More recently, the synergistic Received: January 24, 2016 contributions of bioactive constituents have been scientifically demonstrated. We Revised: February 10, 2016 reviewed the literature concerning medical cannabis and its secondary metabolites, Accepted: February 12, 2016 including fraction and total extracts. Scientific evidence shows that secondary metabolites in cannabis may enhance the positive effects of THC a primary metabolite. Other chemical components (cannabinoid and non-cannabinoid) in cannabis or its extracts may reduce THC-induced anxiety, cholinergic deficits, and immunosuppression; which could increase its therapeutic potential. Particular attention will be placed on non- cannabinoid compounds interactions that could produce synergy with respect to treatment of pain, inflammation, epilepsy, fungal and bacterial infections. -

Endocannabinoids in Body Weight Control
pharmaceuticals Review Endocannabinoids in Body Weight Control Henrike Horn †, Beatrice Böhme †, Laura Dietrich and Marco Koch * Institute of Anatomy, Medical Faculty, University of Leipzig, 04103 Leipzig, Germany; [email protected] (H.H.); [email protected] (B.B.); [email protected] (L.D.) * Correspondence: [email protected]; Tel.: +49-341-97-22047 † These authors contributed equally. Received: 28 April 2018; Accepted: 28 May 2018; Published: 30 May 2018 Abstract: Maintenance of body weight is fundamental to maintain one’s health and to promote longevity. Nevertheless, it appears that the global obesity epidemic is still constantly increasing. Endocannabinoids (eCBs) are lipid messengers that are involved in overall body weight control by interfering with manifold central and peripheral regulatory circuits that orchestrate energy homeostasis. Initially, blocking of eCB signaling by first generation cannabinoid type 1 receptor (CB1) inverse agonists such as rimonabant revealed body weight-reducing effects in laboratory animals and men. Unfortunately, rimonabant also induced severe psychiatric side effects. At this point, it became clear that future cannabinoid research has to decipher more precisely the underlying central and peripheral mechanisms behind eCB-driven control of feeding behavior and whole body energy metabolism. Here, we will summarize the most recent advances in understanding how central eCBs interfere with circuits in the brain that control food intake and -

2 Spice English Presentation
Spice Spice contains no compensatory substances Специи не содержит компенсационные вещества Spice is a mix of herbs (shredded plant material) and manmade chemicals with mind-altering effects. It is often called “synthetic marijuana” because some of the chemicals in it are similar to ones in marijuana; but its effects are sometimes very different from marijuana, and frequently much stronger. It is most often labeled “Not for Human Consumption” and disguised as incense. Eliminationprocess • The synthetic agonists such as THC is fat soluble. • Probably, they are stored as THC in cell membranes. • Some of the chemicals in Spice, however, attach to those receptors more strongly than THC, which could lead to a much stronger and more unpredictable effect. • Additionally, there are many chemicals that remain unidentified in products sold as Spice and it is therefore not clear how they may affect the user. • Moreover, these chemicals are often being changed as the makers of Spice alter them to avoid the products being illegal. • To dissolve the Spice crystals Acetone is used endocannabinoids synhtetic THC cannabinoids CB1 and CB2 agonister Binds to cannabinoidreceptor CB1 CB2 - In the brain -in the immune system Decreased avtivity in the cell ____________________ Maria Ellgren Since some of the compounds have a longer toxic effects compared to naturally THC, as reported: • negative effects that often occur the day after consumption, as a general hangover , but without nausea, mentally slow, confused, distracted, impairment of long and short term memory • Other reports mention the qualitative impairment of cognitive processes and emotional functioning, like all the oxygen leaves the brain. -

In Silico Assessment of Drug-Like Properties of Phytocannabinoids in Cannabis Sativa
EDUCATUM JSMT Vol. 4 No. 2 (2017) ISSN 2289-7070 / eISSN 2462-2451 (1-7) https://ejournal.upsi.edu.my/journal/EDSC In Silico Assessment of Drug-Like Properties of Phytocannabinoids in Cannabis Sativa Shakinaz Desa1*, Asiah Osman2, and Richard Hyslop3 1Department of Biology, Universiti Pendidikan Sultan Idris, Malaysia, 2Natural Product Division, Forest Research Institute Malaysia, 3Department of Chemistry and Biochemistry, University of Northern Colorado, USA *Corresponding author: [email protected] Abstract This study investigated drug-like properties of phytocannabinoids in Cannabis sativa using an in silico study. We report sixteen phytocannabinoids: cannabidiol (CBD), cannabidiolic acid (CBDA), cannabinol (CBN), cannabichromene (CBC), cannabigerol (CBG), cannabicyclol (CBL), cannabivarin (CBV), cannabidivarin (CBDV), cannabichromevarin (CBCV), cannabigerovarin (CBGV), cannabinodiol (CBDL), cannabielsoin (CBE), cannabitriol (CBT), Δ9-tetrahydrocannabinol (Δ9-THC), Δ9-tetrahydrocannabivarin (Δ9-THCV), and Δ8-tetrahydrocannabinol (Δ8-THC). All chemical structures and properties were obtained from PubChem Compound, National Center for Biotechnology Information, U.S. National Library of Medicine. Molinspiration was used for the calculation of molecular properties and bioactivity score. The parameters were molecular weight (MW), number of hydrogen acceptor (HBA), number of hydrogen donor (HBD), partition coefficient (cLogP), polar surface area (PSA) and number of rotatable bonds (NROTB). We predicted bioactivity scores for G Protein-Coupled Receptors (GPCR) ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor and enzyme inhibitor. Lipinski’s rule was used as reference to determine the drug-like properties of the phytocannabinoids. All compounds have MW<500, HBA<10, HBD<5, TPSA<140Å2 and NRTOB<10. Bioactivity score showed an active or moderately active in all compounds. -

The Handbook of Cannabis Therapeutics: from Bench to Bedside
Handbook of Cannabis Therapeutics From Bench to Bedside 9780789030979 Handbook of Cannabis Therapeutics From Bench to Bedside Size: 212 x 152mm Spine size: 26 mm Color pages: Binding: Paperback THE HAWORTH PRESS® Haworth Series in Integrative Healing Ethan Russo Editor The Last Sorcerer: Echoes of the Rainforest by Ethan Russo Professionalism and Ethics in Complementary and Alternative Medicine by John Crellin and Fernando Ania Cannabis and Cannabinoids: Pharmacology, Toxicology, and Therapeutic Potential by Franjo Grotenhermen and Ethan Russo Modern Psychology and Ancient Wisdom: Psychological Healing Practices from the World’s Religious Traditions edited by Sharon G. Mijares Complementary and Alternative Medicine: Clinic Design by Robert A. Roush Herbal Voices: American Herbalism Through the Words of American Herbalists by Anne K. Dougherty The Healing Power of Chinese Herbs and Medicinal Recipes by Joseph P. Hou and Youyu Jin Alternative Therapies in the Treatment of Brain Injury and Neurobehavioral Disorders: A Practical Guide edited by Gregory J. Murrey Handbook of Cannabis Therapeutics: From Bench to Bedside edited by Ethan B. Russo and Franjo Grotenhermen Handbook of Cannabis Therapeutics From Bench to Bedside Ethan B. Russo, MD Franjo Grotenhermen, MD Editors Routledge Taylor &. Francis Croup NEW YORK AND LONDON First Published by The Haworth Press, Inc., 10 Alice Street, Binghamton, NY 13904-1580. Transferred to Digital Printing 2010 by Routledge 270 Madison Ave, New York NY 10016 2 Park Square, Milton Park, Abingdon, Oxon, OX14 4RN For more information on this book or to order, visit http://www.haworthpress.com/store/product.asp?sku=5741 or call 1-800-HAWORTH (800-429-6784) in the United States and Canada or (607) 722-5857 outside the United States and Canada or contact [email protected] © 2006 by The Haworth Press, Inc. -

Cannabinoid Receptor Cannabinoid Receptor
Cannabinoid Receptor Cannabinoid Receptor Cannabinoid receptors are currently classified into three groups: central (CB1), peripheral (CB2) and GPR55, all of which are G-protein-coupled. CB1 receptors are primarily located at central and peripheral nerve terminals. CB2 receptors are predominantly expressed in non-neuronal tissues, particularly immune cells, where they modulate cytokine release and cell migration. Recent reports have suggested that CB2 receptors may also be expressed in the CNS. GPR55 receptors are non-CB1/CB2 receptors that exhibit affinity for endogenous, plant and synthetic cannabinoids. Endogenous ligands for cannabinoid receptors have been discovered, including anandamide and 2-arachidonylglycerol. www.MedChemExpress.com 1 Cannabinoid Receptor Antagonists, Agonists, Inhibitors, Modulators & Activators (S)-MRI-1867 (±)-Ibipinabant Cat. No.: HY-141411A ((±)-SLV319; (±)-BMS-646256) Cat. No.: HY-14791A (S)-MRI-1867 is a peripherally restricted, orally (±)-Ibipinabant ((±)-SLV319) is the racemate of bioavailable dual cannabinoid CB1 receptor and SLV319. (±)-Ibipinabant ((±)-SLV319) is a potent inducible NOS (iNOS) antagonist. (S)-MRI-1867 and selective cannabinoid-1 (CB-1) receptor ameliorates obesity-induced chronic kidney disease antagonist with an IC50 of 22 nM. (CKD). Purity: >98% Purity: 99.93% Clinical Data: No Development Reported Clinical Data: No Development Reported Size: 1 mg, 5 mg Size: 10 mM × 1 mL, 5 mg, 10 mg, 25 mg, 50 mg 2-Arachidonoylglycerol 2-Palmitoylglycerol Cat. No.: HY-W011051 (2-Palm-Gl) Cat. No.: HY-W013788 2-Arachidonoylglycerol is a second endogenous 2-Palmitoylglycerol (2-Palm-Gl), an congener of cannabinoid ligand in the central nervous system. 2-arachidonoylglycerol (2-AG), is a modest cannabinoid receptor CB1 agonist. 2-Palmitoylglycerol also may be an endogenous ligand for GPR119. -

Hemp-Sourced CBD
CLEARING UP CANNABIS: Hemp-Sourced CBD Brooke Weingarden DO Michigan Osteopathic Association Fall Conference 2019 November 10, 2019 1 WHAT DO YOU NEED TO KNOW? What is Hemp-Derived CBD? How is it different than marijuana? Why the political controversy? How is it used? Is it legal in Michigan? What are the known risks/benefits? Research?? Would my patient benefit from it? 2 CBD Cannabinodiol o One of the active ingredients in cannabis, different than THC o No psychoactive effects o Low affinity to the CB1 and CB2 receptors o Enhances 5-HT, Glycine receptors, G couple receptors, effects intracellular calcium, antioxidant effects, anti- inflammatory effects, analgesic o “Charlotte's Web” o Orphan Drug status for treatment resistant childhood seizures (Epidiolex, GW Pharmaceuticals) 3 HEMP VS MARIJUANA: ALL CBD & THC IS NOT THE SAME! THC up to 30%! https://cbdorigin.com/hemp-vs-marijuana/ CBD PHARMACOKINETICS Overall, not well known Majority of data on animals First pass metabolism (Extensive!) Absorption depends on route (IV, oral, inhaled) Half Life: 9-32 hours Low toxicity profile in studies (short term use) Inhibitor of CYP450 (drug-drug interactions) 6 Watch for Drug Interactions! CBD causes potent CYP2C19 inhibition! https://www.ncbi.nlm.nih.gov/pubmed/2331870 8 CYP3A4 Genetics affects the sensitivity to CBD http://profofpot.com/cyp3a4-genetics- cannabinoid-metabolism/ Pharmacytimes.com PROPOSED USES (5) 8 POSSIBLE ADVERSE EFFECTS o Sedative o Psychological o Perception o Motor function o Dependence (9%) o Withdrawals o Stroke o Cerebral blood flow changes o Tachycardia, arrhythmia, MI o Carcinogens I DO NOT RECOMMEND o Slowing of GI system ANY CANNABINOID or o Reproduction CBD PRODUCTS FOR PREGNANT MOTHERS. -

The Phytocannabinoides from Cannabis Sativa L. an Overview
Hop and Medicinal Plants, Year XXVIII, No. 1-2, 2020 ISSN 2360–0179 print, ISSN 2360–0187 electronic THE PHYTOCANNABINOIDES FROM CANNABIS SATIVA L. AN OVERVIEW ONA Andreea Daniela, Sorin MUNTEAN, Leon MUNTEAN* University of Agricultural Sciences and Veterinary Medicine, Faculty of Agriculture, Crop Science Department 3-5 Mănăstur Street, 400372, Cluj-Napoca, Romania *Corresponding author e-mail: [email protected] Abstract: Cannabis sativa is among the first cultivated plants for producing fibres from the stalks and consumption of the seeds. Later, the medicinal properties were discovered. The oldest written record of using cannabis for the medicinal and recreational purposes is from China. Over the time many experimental researches were carried out that emphasize the importance of cannabinoids for human health. The present paper focused to present an overview of the main cannabinoids synthetized in cannabis plant and their medicinal and therapeutic effects. Cannabis has a complex chemical composition including besides cannabinoids (over 100), also terpenoids, sugars, alkaloids, quinones and stilbenoids. Of major importance are two compounds tetrahydrocannabinol (THC) and cannabidiol (CBD). Over the time different researches pointed out the beneficial effect of cannabinoids on a wide range of diseases such as epilepsy, neurological diseases, mental conditions like anxiety and depression, chronic pains and certain forms of cancer. In addition to the important advantages it must be also paid attention to the psychoactive action of the certain cannabinoids that may produce cognitive disturbances, attention disorders, sever panic, can affect the short-term memory, and slow the reaction time. This deficits induced usually by THC can be moderate by using CBD which reduce the psychoactive effect. -
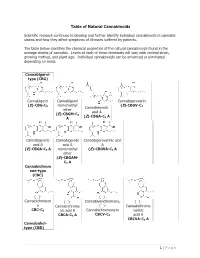
Table of Natural Cannabinoids
Table of Natural Cannabinoids Scientific research continues to develop and further identify individual cannabinoids in cannabis strains and how they affect symptoms of illnesses suffered by patients. The table below identifies the chemical properties of the natural cannabinoids found in the average strains of cannabis. Levels of each of these chemicals will vary with varietal strain, growing method, and plant age. Individual cannabinoids can be enhanced or eliminated depending on need. Cannabigerol- type (CBG) Cannabigerol Cannabigerol Cannabigerovarin (E)-CBG-C monomethyl (E)-CBGV-C 5 Cannabinerolic 3 ether acid A (E)-CBGM-C 5 (Z)-CBGA-C A A 5 Cannabigerolic Cannabigerolic Cannabigerovarinic acid acid A acid A A (E)-CBGA-C5 A monomethyl (E)-CBGVA-C3 A ether (E)-CBGAM- C5 A Cannabichrom ene-type (CBC) (±)- (±)- Cannabichromen (±)- Cannabivarichromene, (±)- e Cannabichrome (±)- Cannabichrome CBC-C5 nic acid A Cannabichromevarin varinic CBCA-C5 A CBCV-C3 acid A CBCVA-C3 A Cannabidiol- type (CBD) 1 | Page (−)-Cannabidiol Cannabidiol Cannabidiol-C4 (−)- Cannabidiorc CBD-C5 momomethyl CBD-C4 Cannabidivarin ol ether CBDV-C3 CBD-C1 CBDM-C5 Cannabidiolic Cannabidivarini acid c acid CBDA-C5 CBDVA-C3 Cannabinodiol- type (CBND) Cannabinodiol Cannabinodivar CBND-C5 in CBND-C3 Tetrahydrocan nabinol-type (THC) 9 9 9 Δ - Δ - Δ - Δ9- Tetrahydrocanna Tetrahydrocan Tetrahydrocannabivarin 9 Tetrahydrocan binol nabinol-C4 Δ -THCV-C3 9 9 nabiorcol Δ -THC-C5 Δ -THC-C4 9 Δ -THCO-C1 9 9 Δ -Tetrahydro- Δ9-Tetrahydro- Δ -Tetrahydro- Δ9-Tetrahydro- cannabinolic