Dissertation
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Control Engineering Perspective on Genome-Scale Metabolic Modeling
Control Engineering Perspective on Genome-Scale Metabolic Modeling by Andrew Louis Damiani A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama December 12, 2015 Key words: Scheffersomyces stipitis, Flux Balance Analysis, Genome-scale metabolic models, System Identification Framework, Model Validation, Phenotype Phase Plane Analysis Copyright 2015 by Andrew Damiani Approved by Jin Wang, Chair, Associate Professor of Chemical Engineering Q. Peter He, Associate Professor of Chemical Engineering, Tuskegee University Thomas W. Jeffries, Professor of Bacteriology, Emeritus; University of Wisconsin-Madison Allan E. David, Assistant Professor of Chemical Engineering Yoon Y. Lee, Professor of Chemical Engineering Abstract Fossil fuels impart major problems on the global economy and have detrimental effects to the environment, which has caused a world-wide initiative of producing renewable fuels. Lignocellulosic bioethanol for renewable energy has recently gained attention, because it can overcome the limitations that first generation biofuels impose. Nonetheless, in order to have this process commercialized, the biological conversion of pentose sugars, mainly xylose, needs to be improved. Scheffersomyces stipitis has a physiology that makes it a valuable candidate for lignocellulosic bioethanol production, and lately has provided genes for designing recombinant Saccharomyces cerevisiae. In this study, a system biology approach was taken to understand the relationship of the genotype to phenotype, whereby genome-scale metabolic models (GSMMs) are used in conjunction with constraint-based modeling. The major restriction of GSMMs is having an accurate methodology for validation and evaluation. This is due to the size and complexity of the models. -

Open Matthew R Moreau Ph.D. Dissertation Finalfinal.Pdf
The Pennsylvania State University The Graduate School Department of Veterinary and Biomedical Sciences Pathobiology Program PATHOGENOMICS AND SOURCE DYNAMICS OF SALMONELLA ENTERICA SEROVAR ENTERITIDIS A Dissertation in Pathobiology by Matthew Raymond Moreau 2015 Matthew R. Moreau Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2015 The Dissertation of Matthew R. Moreau was reviewed and approved* by the following: Subhashinie Kariyawasam Associate Professor, Veterinary and Biomedical Sciences Dissertation Adviser Co-Chair of Committee Bhushan M. Jayarao Professor, Veterinary and Biomedical Sciences Dissertation Adviser Co-Chair of Committee Mary J. Kennett Professor, Veterinary and Biomedical Sciences Vijay Kumar Assistant Professor, Department of Nutritional Sciences Anthony Schmitt Associate Professor, Veterinary and Biomedical Sciences Head of the Pathobiology Graduate Program *Signatures are on file in the Graduate School iii ABSTRACT Salmonella enterica serovar Enteritidis (SE) is one of the most frequent common causes of morbidity and mortality in humans due to consumption of contaminated eggs and egg products. The association between egg contamination and foodborne outbreaks of SE suggests egg derived SE might be more adept to cause human illness than SE from other sources. Therefore, there is a need to understand the molecular mechanisms underlying the ability of egg- derived SE to colonize the chicken intestinal and reproductive tracts and cause disease in the human host. To this end, the present study was carried out in three objectives. The first objective was to sequence two egg-derived SE isolates belonging to the PFGE type JEGX01.0004 to identify the genes that might be involved in SE colonization and/or pathogenesis. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Product Sheet Info
Master Clone List for NR-19279 ® Vibrio cholerae Gateway Clone Set, Recombinant in Escherichia coli, Plates 1-46 Catalog No. NR-19279 Table 1: Vibrio cholerae Gateway® Clones, Plate 1 (NR-19679) Clone ID Well ORF Locus ID Symbol Product Accession Position Length Number 174071 A02 367 VC2271 ribD riboflavin-specific deaminase NP_231902.1 174346 A03 336 VC1877 lpxK tetraacyldisaccharide 4`-kinase NP_231511.1 174354 A04 342 VC0953 holA DNA polymerase III, delta subunit NP_230600.1 174115 A05 388 VC2085 sucC succinyl-CoA synthase, beta subunit NP_231717.1 174310 A06 506 VC2400 murC UDP-N-acetylmuramate--alanine ligase NP_232030.1 174523 A07 132 VC0644 rbfA ribosome-binding factor A NP_230293.2 174632 A08 322 VC0681 ribF riboflavin kinase-FMN adenylyltransferase NP_230330.1 174930 A09 433 VC0720 phoR histidine protein kinase PhoR NP_230369.1 174953 A10 206 VC1178 conserved hypothetical protein NP_230823.1 174976 A11 213 VC2358 hypothetical protein NP_231988.1 174898 A12 369 VC0154 trmA tRNA (uracil-5-)-methyltransferase NP_229811.1 174059 B01 73 VC2098 hypothetical protein NP_231730.1 174075 B02 82 VC0561 rpsP ribosomal protein S16 NP_230212.1 174087 B03 378 VC1843 cydB-1 cytochrome d ubiquinol oxidase, subunit II NP_231477.1 174099 B04 383 VC1798 eha eha protein NP_231433.1 174294 B05 494 VC0763 GTP-binding protein NP_230412.1 174311 B06 314 VC2183 prsA ribose-phosphate pyrophosphokinase NP_231814.1 174603 B07 108 VC0675 thyA thymidylate synthase NP_230324.1 174474 B08 466 VC1297 asnS asparaginyl-tRNA synthetase NP_230942.2 174933 B09 198 -

The Metabolic Building Blocks of a Minimal Cell Supplementary
The metabolic building blocks of a minimal cell Mariana Reyes-Prieto, Rosario Gil, Mercè Llabrés, Pere Palmer and Andrés Moya Supplementary material. Table S1. List of enzymes and reactions modified from Gabaldon et. al. (2007). n.i.: non identified. E.C. Name Reaction Gil et. al. 2004 Glass et. al. 2006 number 2.7.1.69 phosphotransferase system glc + pep → g6p + pyr PTS MG041, 069, 429 5.3.1.9 glucose-6-phosphate isomerase g6p ↔ f6p PGI MG111 2.7.1.11 6-phosphofructokinase f6p + atp → fbp + adp PFK MG215 4.1.2.13 fructose-1,6-bisphosphate aldolase fbp ↔ gdp + dhp FBA MG023 5.3.1.1 triose-phosphate isomerase gdp ↔ dhp TPI MG431 glyceraldehyde-3-phosphate gdp + nad + p ↔ bpg + 1.2.1.12 GAP MG301 dehydrogenase nadh 2.7.2.3 phosphoglycerate kinase bpg + adp ↔ 3pg + atp PGK MG300 5.4.2.1 phosphoglycerate mutase 3pg ↔ 2pg GPM MG430 4.2.1.11 enolase 2pg ↔ pep ENO MG407 2.7.1.40 pyruvate kinase pep + adp → pyr + atp PYK MG216 1.1.1.27 lactate dehydrogenase pyr + nadh ↔ lac + nad LDH MG460 1.1.1.94 sn-glycerol-3-phosphate dehydrogenase dhp + nadh → g3p + nad GPS n.i. 2.3.1.15 sn-glycerol-3-phosphate acyltransferase g3p + pal → mag PLSb n.i. 2.3.1.51 1-acyl-sn-glycerol-3-phosphate mag + pal → dag PLSc MG212 acyltransferase 2.7.7.41 phosphatidate cytidyltransferase dag + ctp → cdp-dag + pp CDS MG437 cdp-dag + ser → pser + 2.7.8.8 phosphatidylserine synthase PSS n.i. cmp 4.1.1.65 phosphatidylserine decarboxylase pser → peta PSD n.i. -

Structure and Mechanism of a Eukaryotic Fmn Adenylyltransferase
STRUCTURE AND MECHANISM OF A EUKARYOTIC FMN ADENYLYLTRANSFERASE Approved by supervisory committee ____________________________________ Hong Zhang, Ph.D. (Mentor) ____________________________________ Diana Tomchick, Ph.D. (Committee Chair) ____________________________________ Kevin Gardner, Ph.D. ____________________________________ Betsy Goldsmith, Ph.D. This thesis is dedicated to my family. STRUCTURE AND MECHANISM OF A EUKARYOTIC FMN ADENYLYLTRANSFERASE by CARLOS HUERTA JR. DISSERTATION Presented to the Faculty of the Graduate School of Biomedical Sciences The University of Texas Southwestern Medical Center at Dallas In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY The University of Texas Southwestern Medical Center at Dallas Dallas, Texas December, 2009 Copyright by CARLOS HUERTA JR., 2009 All Rights Reserved ACKNOWLEDGEMENTS There are many individuals that contributed to my graduate education and I would like to thank a few of them. First, I would like to honor my mentor, Dr. Hong Zhang. Dr. Zhang’s support and encouragement was essential to the completion of my graduate education. Dr. Zhang’s compassion to X-ray crystallography and biological science was fundamental in transforming me into a structural biologist. I would also like to thank all past and current members of the Zhang laboratory. In particular, I would like to thank Dr. Nian Huang and Dr. Darek Martynowski for discussions in structure refinement and modeling, and Marcelo Raines for teaching me protein purification and crystallization. A special acknowledgement goes to Dr. Dominika Borek for teaching me how to solve my first protein structure and her support through-out my graduate education. My graduate thesis would not be possible without the guidance and understanding of my dissertation committee members Dr. -

The Action Mechanism of Prokaryotic FAD Synthetases As a Potential Drug Target for the Treatment of Bacterial Infections
2018 112 María Sebastián Valverde The action mechanism of prokaryotic FAD synthetases as a potential drug target for the treatment of bacterial infections Departamento Bioquímica y Biología Molecular y Celular Director/es MEDINA TRULLENQUE, MILAGROS Reconocimiento – NoComercial – © Universidad de Zaragoza SinObraDerivada (by-nc-nd): No se Servicio de Publicaciones permite un uso comercial de la obra original ni la generación de obras derivadas. ISSN 2254-7606 Tesis Doctoral THE ACTION MECHANISM OF PROKARYOTIC FAD SYNTHETASES AS A POTENTIAL DRUG TARGET FOR THE TREATMENT OF BACTERIAL INFECTIONS Autor María Sebastián Valverde Director/es MEDINA TRULLENQUE, MILAGROS UNIVERSIDAD DE ZARAGOZA Bioquímica y Biología Molecular y Celular 2018 Repositorio de la Universidad de Zaragoza – Zaguan http://zaguan.unizar.es Abstract 1 Resumen 7 Chapter I: Introduction 13 1. The need of new antibacterial targets 15 1.1. From the beginnings to the golden era in the development of antibiotics 15 1.2. The resistance era put in check our effectiveness against pathogens 16 1.3. What makes a drug target good? 17 1.4. Proteins involved in flavins synthesis are promising drug targets 18 2. Flavins and flavoproteins: past, present and properties 20 2.1. A brief history of flavins and flavoproteins 20 2.2. Flavins are heterocyclic molecules 21 2.3. Flavins exhibit versatile redox properties 23 2.4. Flavins show useful spectroscopic properties 24 2.4.1. Absorption in the UV depends on the redox state 24 2.4.2. Flavins are fluorescent compounds 26 2.5. Biosynthesis of FMN and FAD in different organisms 26 3. FMN and FAD biosynthesis in different types of organisms 27 3.1. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Genome-Scale Metabolic Network Analysis and Drug Targeting of Multi-Drug Resistant Pathogen Acinetobacter Baumannii AYE
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2017 Electronic Supplementary Information (ESI) for Molecular BioSystems Genome-scale metabolic network analysis and drug targeting of multi-drug resistant pathogen Acinetobacter baumannii AYE Hyun Uk Kim, Tae Yong Kim and Sang Yup Lee* E-mail: [email protected] Supplementary Table 1. Metabolic reactions of AbyMBEL891 with information on their genes and enzymes. Supplementary Table 2. Metabolites participating in reactions of AbyMBEL891. Supplementary Table 3. Biomass composition of Acinetobacter baumannii. Supplementary Table 4. List of 246 essential reactions predicted under minimal medium with succinate as a sole carbon source. Supplementary Table 5. List of 681 reactions considered for comparison of their essentiality in AbyMBEL891 with those from Acinetobacter baylyi ADP1. Supplementary Table 6. List of 162 essential reactions predicted under arbitrary complex medium. Supplementary Table 7. List of 211 essential metabolites predicted under arbitrary complex medium. AbyMBEL891.sbml Genome-scale metabolic model of Acinetobacter baumannii AYE, AbyMBEL891, is available as a separate file in the format of Systems Biology Markup Language (SBML) version 2. Supplementary Table 1. Metabolic reactions of AbyMBEL891 with information on their genes and enzymes. Highlighed (yellow) reactions indicate that they are not assigned with genes. No. Metabolism EC Number ORF Reaction Enzyme R001 Glycolysis/ Gluconeogenesis 5.1.3.3 ABAYE2829 -
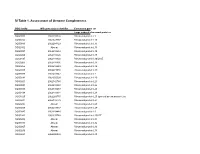
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Supplementary Material
Supplementary Material TABLE S1: List of FSFs that were likely acquired via HGT in the AB, AE, and BE taxonomic groups. Both the SCOP and alphanumeric identifiers were used to define FSFs. HGT direction was inferred from fdifference, which is the difference in f-values between fformer (e.g. A in the AB taxonomic group) and flatter (B in AB). Taxonomic SCOP HGT FSF Id FSF name f f f group Id former latter difference direction Sulfolobus fructose- AB 111249 d.280.1 1,6-bisphosphatase- 0.7143 0.0353 0.679 A to B like tRNA-intron endonuclease N- AE 55267 d.75.1 0.9429 0.0154 0.9274 A to E terminal domain- like AN1-like Zinc AE 118310 g.80.1 0.3143 0.9498 -0.6355 E to A finger Mitochondrial AE 47157 a.23.4 import receptor 0.0143 0.7066 -0.6923 E to A subunit Tom20 AE 55418 d.86.1 eIF4e-like 0.1286 1 -0.8714 E to A Supernatant protein AE 101576 b.132.1 factor (SPF), C- 0.0143 0.8996 -0.8853 E to A terminal domain Eukaryotic DNA topoisomerase I, N- AE 56741 e.15.1 0.0143 0.9498 -0.9355 E to A terminal DNA- binding fragment AE 109993 a.222.1 VPS9 domain 0.0143 0.9846 -0.9703 E to A Transcription factor BE 69705 d.202.1 NusA, N-terminal 0.9954 0.0039 0.9915 B to E domain BE 48295 a.4.12 TrpR-like 0.9831 0.0039 0.9793 B to E Small protein B BE 74982 b.111.1 0.9985 0.0347 0.9637 B to E (SmpB) N-terminal domain BE 48024 a.81.1 0.9877 0.027 0.9607 B to E of DnaB helicase C-terminal domain BE 47789 a.60.3 of RNA polymerase 0.9954 0.0502 0.9452 B to E alpha subunit GreA transcript BE 46557 a.2.1 cleavage protein, N- 0.9555 0.0116 0.9439 B to E terminal -

Physiological Models of Geobacter Sulfurreducens and Desulfobacter Postgatei to Understand Uranium Remediation in Subsurface Systems
University of Massachusetts Amherst ScholarWorks@UMass Amherst Doctoral Dissertations Dissertations and Theses November 2014 PHYSIOLOGICAL MODELS OF GEOBACTER SULFURREDUCENS AND DESULFOBACTER POSTGATEI TO UNDERSTAND URANIUM REMEDIATION IN SUBSURFACE SYSTEMS Roberto Orellana University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/dissertations_2 Part of the Environmental Microbiology and Microbial Ecology Commons, and the Microbial Physiology Commons Recommended Citation Orellana, Roberto, "PHYSIOLOGICAL MODELS OF GEOBACTER SULFURREDUCENS AND DESULFOBACTER POSTGATEI TO UNDERSTAND URANIUM REMEDIATION IN SUBSURFACE SYSTEMS" (2014). Doctoral Dissertations. 278. https://doi.org/10.7275/5822237.0 https://scholarworks.umass.edu/dissertations_2/278 This Open Access Dissertation is brought to you for free and open access by the Dissertations and Theses at ScholarWorks@UMass Amherst. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected]. PHYSIOLOGICAL MODELS OF GEOBACTER SULFURREDUCENS AND DESULFOBACTER POSTGATEI TO UNDERSTAND URANIUM REMEDIATION IN SUBSURFACE SYSTEMS A Dissertation Presented by ROBERTO ORELLANA ROMAN Submitted to the Graduate School of the University of Massachusetts Amherst in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY September 2014 Microbiology Department © Copyright by Roberto Orellana Roman 2014 All Rights