COPEIA June 12
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Ecology and Evolutionary History of Two Musk Turtles in the Southeastern United States
The University of Southern Mississippi The Aquila Digital Community Dissertations Spring 2020 The Ecology and Evolutionary History of Two Musk Turtles in the Southeastern United States Grover Brown Follow this and additional works at: https://aquila.usm.edu/dissertations Part of the Genetics Commons Recommended Citation Brown, Grover, "The Ecology and Evolutionary History of Two Musk Turtles in the Southeastern United States" (2020). Dissertations. 1762. https://aquila.usm.edu/dissertations/1762 This Dissertation is brought to you for free and open access by The Aquila Digital Community. It has been accepted for inclusion in Dissertations by an authorized administrator of The Aquila Digital Community. For more information, please contact [email protected]. THE ECOLOGY AND EVOLUTIONARY HISTORY OF TWO MUSK TURTLES IN THE SOUTHEASTERN UNITED STATES by Grover James Brown III A Dissertation Submitted to the Graduate School, the College of Arts and Sciences and the School of Biological, Environmental, and Earth Sciences at The University of Southern Mississippi in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Approved by: Brian R. Kreiser, Committee Co-Chair Carl P. Qualls, Committee Co-Chair Jacob F. Schaefer Micheal A. Davis Willian W. Selman II ____________________ ____________________ ____________________ Dr. Brian R. Kreiser Dr. Jacob Schaefer Dr. Karen S. Coats Committee Chair Director of School Dean of the Graduate School May 2020 COPYRIGHT BY Grover James Brown III 2020 Published by the Graduate School ABSTRACT Turtles are among one of the most imperiled vertebrate groups on the planet with more than half of all species worldwide listed as threatened, endangered or extinct by the International Union of the Conservation of Nature. -

A Molecular Phylogeny for Marine Turtles: Trait Mapping, Rate
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 5574-5577, June 1993 Evolution A molecular phylogeny for marine turtles: Trait mapping, rate assessment, and conservation relevance (conservation genetlcs/cytochrome b/molecular systematics/mitochondrial DNA) BRIAN W. BOWEN*, WILLIAM S. NELSON, AND JOHN C. AVISE Department of Genetics, University of Georgia, Athens, GA 30602 Contributed by John C. Avise, March 25, 1993 ABSTRACT Nucleotide sequences from the cytochrome b Table 1. A recent taxonomy for marine turtles, with problematic gene of mitochondrial DNA were employed to resolve phylo- areas indicated by parenthetical questions genetic controversies and to assess molecular evolutionary rates Order Testudines-all extant turtles, freshwater, terrestrial, and in marine turtles (Chelonioidea). Findings of special relevance marine to conservation biology include discovery of a distant relation- Family Dermochelyidae-Dermochelys coriacea (leatherback) ship betweenNatatorand other chelonild species, the paraphyly (Are marine turtles monophyletic? Where does this species lie ofChelonia mydas with respect to Chelonia agassizi, and genetic with regard to broader turtle phylogeny?) distinctiveness of Lepidochelys kempi from Lepidochelys oliva- Family Cheloniidae cea. A longstanding debate in evolutionary ecology was re- Tribe Chelonini solved by phylogenetic mapping of dietary habits, which indi- Genus Chelonia-C. mydas (green) cates that the spongivore Ereinochelys imbricata evolved from C. agassizi (black) a carnivorous rather than a herbivorous ancestor. Sequence (Are these two forms distinct species?) divergences at intergeneric and interfamilial levels, when as- Genus Natator-N. depressus (flatback) sessed against fossil-based separation times, support previous (Is this species a close ally of the green turtle, or perhaps suggestions (from microevolutionary comparisons) that mito- allied more closely to members of the Carettini?) chondrial DNA in marine turtles evolves much more slowly Tribe Carettini than under the "conventional" vertebrate clock. -
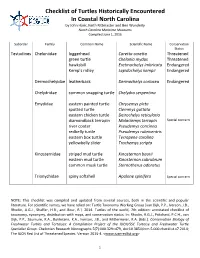
N.C. Turtles Checklist
Checklist of Turtles Historically Encountered In Coastal North Carolina by John Hairr, Keith Rittmaster and Ben Wunderly North Carolina Maritime Museums Compiled June 1, 2016 Suborder Family Common Name Scientific Name Conservation Status Testudines Cheloniidae loggerhead Caretta caretta Threatened green turtle Chelonia mydas Threatened hawksbill Eretmochelys imbricata Endangered Kemp’s ridley Lepidochelys kempii Endangered Dermochelyidae leatherback Dermochelys coriacea Endangered Chelydridae common snapping turtle Chelydra serpentina Emydidae eastern painted turtle Chrysemys picta spotted turtle Clemmys guttata eastern chicken turtle Deirochelys reticularia diamondback terrapin Malaclemys terrapin Special concern river cooter Pseudemys concinna redbelly turtle Pseudemys rubriventris eastern box turtle Terrapene carolina yellowbelly slider Trachemys scripta Kinosternidae striped mud turtle Kinosternon baurii eastern mud turtle Kinosternon subrubrum common musk turtle Sternotherus odoratus Trionychidae spiny softshell Apalone spinifera Special concern NOTE: This checklist was compiled and updated from several sources, both in the scientific and popular literature. For scientific names, we have relied on: Turtle Taxonomy Working Group [van Dijk, P.P., Iverson, J.B., Rhodin, A.G.J., Shaffer, H.B., and Bour, R.]. 2014. Turtles of the world, 7th edition: annotated checklist of taxonomy, synonymy, distribution with maps, and conservation status. In: Rhodin, A.G.J., Pritchard, P.C.H., van Dijk, P.P., Saumure, R.A., Buhlmann, K.A., Iverson, J.B., and Mittermeier, R.A. (Eds.). Conservation Biology of Freshwater Turtles and Tortoises: A Compilation Project of the IUCN/SSC Tortoise and Freshwater Turtle Specialist Group. Chelonian Research Monographs 5(7):000.329–479, doi:10.3854/crm.5.000.checklist.v7.2014; The IUCN Red List of Threatened Species. -

Gopher Tortoise Demographic Responses to a Novel Disturbance Regime
The Journal of Wildlife Management 1–10; 2019; DOI: 10.1002/jwmg.21774 Research Article Gopher Tortoise Demographic Responses to a Novel Disturbance Regime HUNTER J. HOWELL ,1,2 Department of Biology, University of Miami, 1301 Memorial Drive, Coral Gables, FL 33146, USA BETSIE B. ROTHERMEL,2 Archbold Biological Station, 123 Main Drive, Venus, FL 33960, USA K. NICOLE WHITE, Daniel B. Warnell School of Forestry and Natural Resources, University of Georgia, 180 E Green Street, Athens, GA 30602, USA CHRISTOPHER A. SEARCY, Department of Biology, University of Miami, 1301 Memorial Drive, Coral Gables, FL 33146, USA ABSTRACT The long‐term viability of gopher tortoise (Gopherus polyphemus) populations is jeopardized by increased urbanization and habitat degradation owing to fire suppression. Because the species’ remaining natural habitats in the southeastern United States exist within a mosaic of anthropogenic land uses, it is important to understand demographic responses to contrasting land uses and habitat management regimes. We examined differences in demographic parameters among fire‐suppressed sandhill, restored sandhill, and former sandhill (i.e., ruderal) land use‐land cover (LULC) types at Archbold Biological Station in south‐ central Florida, USA. Using Program MARK, we estimated population size, and sex‐specific and LULC‐specific survivorship based on 6 years of mark‐recapture data. We also analyzed individual growth trajectories and clutch sizes to determine whether growth rates or reproductive output differed among LULC types. Tortoises in an open, ruderal field occurred at a higher density (7.79/ha) than in adjacent restored (1.43/ha) or fire‐suppressed (0.40/ha) sandhill. Despite this higher density, both adult survivorship and body size were significantly higher in the ruderal field. -

The Conservation Biology of Tortoises
The Conservation Biology of Tortoises Edited by Ian R. Swingland and Michael W. Klemens IUCN/SSC Tortoise and Freshwater Turtle Specialist Group and The Durrell Institute of Conservation and Ecology Occasional Papers of the IUCN Species Survival Commission (SSC) No. 5 IUCN—The World Conservation Union IUCN Species Survival Commission Role of the SSC 3. To cooperate with the World Conservation Monitoring Centre (WCMC) The Species Survival Commission (SSC) is IUCN's primary source of the in developing and evaluating a data base on the status of and trade in wild scientific and technical information required for the maintenance of biological flora and fauna, and to provide policy guidance to WCMC. diversity through the conservation of endangered and vulnerable species of 4. To provide advice, information, and expertise to the Secretariat of the fauna and flora, whilst recommending and promoting measures for their con- Convention on International Trade in Endangered Species of Wild Fauna servation, and for the management of other species of conservation concern. and Flora (CITES) and other international agreements affecting conser- Its objective is to mobilize action to prevent the extinction of species, sub- vation of species or biological diversity. species, and discrete populations of fauna and flora, thereby not only maintain- 5. To carry out specific tasks on behalf of the Union, including: ing biological diversity but improving the status of endangered and vulnerable species. • coordination of a programme of activities for the conservation of biological diversity within the framework of the IUCN Conserva- tion Programme. Objectives of the SSC • promotion of the maintenance of biological diversity by monitor- 1. -

Relative Abundance, Population Structure, and Habitat
University of Missouri, St. Louis IRL @ UMSL Theses Graduate Works 11-18-2010 Relative Abundance, Population Structure, and Habitat Utilization of the Alligator Snapping Turtle (Macrochelys temminckii) and Eastern Snapping Turtle (Chelydra serpentina) in southeastern Missouri Timothy Charles Lescher University of Missouri-St. Louis, [email protected] Follow this and additional works at: http://irl.umsl.edu/thesis Recommended Citation Lescher, Timothy Charles, "Relative Abundance, Population Structure, and Habitat Utilization of the Alligator Snapping Turtle (Macrochelys temminckii) and Eastern Snapping Turtle (Chelydra serpentina) in southeastern Missouri" (2010). Theses. 44. http://irl.umsl.edu/thesis/44 This Thesis is brought to you for free and open access by the Graduate Works at IRL @ UMSL. It has been accepted for inclusion in Theses by an authorized administrator of IRL @ UMSL. For more information, please contact [email protected]. Relative Abundance, Population Structure, and Habitat Utilization of the Alligator Snapping Turtle (Macrochelys temminckii) and Eastern Snapping Turtle (Chelydra serpentina) in southeastern Missouri Timothy C. Lescher B.S., Biology, University of Missouri-Columbia, 2001 A Thesis Submitted to the Graduate School at the University of Missouri-Saint Louis in partial fulfillment of the requirements for the degree Master’s of Science in Biology November 2010 Advisory Committee Zuleyma Tang-Martinez, Ph.D. Chair Jeffrey T. Briggler, Ph.D. Bette A. Loiselle, Ph.D. Copyright, Timothy C. Lescher, 2010 1 TABLE -

A Case of Inversion and Duplication Involving Constitutive Heterochromatin
Genetics and Molecular Biology, 36, 3, 353-356 (2013) Copyright © 2013, Sociedade Brasileira de Genética. Printed in Brazil www.sbg.org.br Short Communication Cytogenetic comparison of Podocnemis expansa and Podocnemis unifilis: A case of inversion and duplication involving constitutive heterochromatin Ricardo José Gunski1, Isabel Souza Cunha2, Tiago Marafiga Degrandi1, Mario Ledesma3 and Analía Del Valle Garnero1 1Universidade Federal do Pampa, Campus São Gabriel, São Gabriel, RS, Brazil. 2Secretaria Municipal de Meio Ambiente e Turismo, São Desidério, BA, Brazil. 3Parque Ecológico “El Puma”, Candelaria, Argentina. Abstract Podocnemis expansa and P. unifilis present 2n = 28 chromosomes, a diploid number similar to those observed in other species of the genus. The aim of this study was to characterize these two species using conventional staining and differential CBG-, GTG and Ag-NOR banding. We analyzed specimens of P. expansa and P. unifilis from the state of Tocantins (Brazil), in which we found a 2n = 28 and karyotypes differing in the morphology of the 13th pair, which was submetacentric in P. expansa and telocentric in P. unifilis. The CBG-banding patterns revealed a heterochromatic block in the short arm of pair 13 of P. expansa and an interstitial one in pair 13 of P. unifilis, suggest- ing a pericentric inversion. Pair 14 of P. unifilis showed an insterstitial band in the long arm that was absent in P. expansa, suggesting a duplication in this region. Ag-NORs were observed in the first chromosome pair of both spe- cies and was associated to a secondary constriction and heterochromatic blocks. Keywords: chromosome banding, Ag-NORs, karyotype, chelonids. -

Dermatemys Mawii (The Hicatee, Tortuga Blanca, Or Central American River Turtle): a Working Bibliography
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/325206006 Dermatemys mawii (The Hicatee, Tortuga Blanca, or Central American River Turtle): A Working Bibliography Article · May 2018 CITATIONS READS 0 521 6 authors, including: Venetia Briggs Sergio C. Gonzalez University of Florida University of Florida 16 PUBLICATIONS 133 CITATIONS 9 PUBLICATIONS 39 CITATIONS SEE PROFILE SEE PROFILE Thomas Rainwater Clemson University 143 PUBLICATIONS 1,827 CITATIONS SEE PROFILE Some of the authors of this publication are also working on these related projects: Crocodiles in the Everglades View project Dissertation: The role of habitat expansion on amphibian community structure View project All content following this page was uploaded by Sergio C. Gonzalez on 17 May 2018. The user has requested enhancement of the downloaded file. 2018 Endangered and ThreatenedCaribbean Species Naturalist of the Caribbean Region Special Issue No. 2 V. Briggs-Gonzalez, S.C. Gonzalez, D. Smith, K. Allen, T.R. Rainwater, and F.J. Mazzotti 2018 CARIBBEAN NATURALIST Special Issue No. 2:1–22 Dermatemys mawii (The Hicatee, Tortuga Blanca, or Central American River Turtle): A Working Bibliography Venetia Briggs-Gonzalez1,*, Sergio C. Gonzalez1, Dustin Smith2, Kyle Allen1, Thomas R. Rainwater3, and Frank J. Mazzotti1 Abstract - Dermatemys mawii (Central American River Turtle), locally known in Belize as the “Hicatee” and in Guatemala and Mexico as Tortuga Blanca, is a large, highly aquatic freshwater turtle that has been extirpated from much of its historical range of southern Mex- ico, northern Guatemala, and lowland Belize. Throughout its restricted range, Dermatemys has been intensely harvested for its meat and eggs and sold in local markets. -

Turtles, All Marine Turtles, Have Been Documented Within the State’S Borders
Turtle Only four species of turtles, all marine turtles, have been documented within the state’s borders. Terrestrial and freshwater aquatic species of turtles do not occur in Alaska. Marine turtles are occasional visitors to Alaska’s Gulf Coast waters and are considered a natural part of the state’s marine ecosystem. Between 1960 and 2007 there were 19 reports of leatherback sea turtles (Dermochelys coriacea), the world’s largest turtle. There have been 15 reports of Green sea turtles (Chelonia mydas). The other two are extremely rare, there have been three reports of Olive ridley sea turtles (Lepidochelys olivacea) and two reports of loggerhead sea turtles (Caretta caretta). Currently, all four species are listed as threatened or endangered under the U.S. Endangered Species Act. Prior to 1993, Alaska marine turtle sightings were mostly of live leatherback sea turtles; since then most observations have been of green sea turtle carcasses. At present, it is not possible to determine if this change is related to changes in oceanographic conditions, perhaps as the result of global warming, or to changes in the overall population size and distribution of these species. General description: Marine turtles are large, tropical/subtropical, thoroughly aquatic reptiles whose forelimbs or flippers are specially modified for swimming and are considerably larger than their hind limbs. Movements on land are awkward. Except for occasional basking by both sexes and egg-laying by females, turtles rarely come ashore. Turtles are among the longest-lived vertebrates. Although their age is often exaggerated, they probably live 50 to 100 years. Of the five recognized species of marine turtles, four (including the green sea turtle) belong to the family Cheloniidae. -

Harvested Testudines
Testudines HARVESTED – Is the species collected by humans? Species Common Name Harvested Cheloniidae sea turtles Caretta c. caretta Atlantic loggerhead Unk Chelonia m. mydas Atlantic green turtle YF Economically Chelonia mydas is the most important reptile in the world. Its flesh and its eggs serve as an important source of protein in many third-world nations where protein is scarce (Ernst et al. 1994) Eretmochelys i. imbricata Atlantic hawksbill YF hawksbill flesh and eggs are eaten in many parts of its range (Ernst et al. 1994); YC throughout its range it is hunted for the plates of its shells (Ernst et al. 1994) Lepidochelys kempii Kemp’s ridley or Atlantic ridley YF egg poaching caused the initial decline [in population size] (Delikat 1981; Hall et al. 1983; Lutz and Lutcavage 1989; Ross et al. 1989; Thompson 1989; National Research Council 1990; Caillouet et al. 1991) Dermochelyidae leatherback sea turtles Dermochelys c. coriacea Atlantic leatherback YF gathering the eggs for both commerce and personal consumption (Ernst et al. 1994) Chelydridae snapping turtles Chelydra s. serpentina eastern snapping turtle YF the flesh of the snapping turtle is tasty and eaten throughout the range…fresh eggs are edible if fried (Ernst et al. 1994) Emydidae pond turtles Chrysemys p. picta eastern painted turtle Unk Chrysemys p. marginata midland painted turtle Unk Clemmys guttata spotted turtle YP (Lovich and Jaworski 1988; Lovich 1989; J. Harding, pers. comm.) Clemmys insculpta wood turtle YP (Ernst et al. 1994) Clemmys muhlenbergii bog turtle YP (D.E. Collins 1990); YF eaten by people (Ernst et al. 1994) Deirochelys r. -

The Staurotypus Turtles and Aves Share the Same Origin of Sex Chromosomes but Evolved Different Types of Heterogametic Sex Determination
The Staurotypus Turtles and Aves Share the Same Origin of Sex Chromosomes but Evolved Different Types of Heterogametic Sex Determination Taiki Kawagoshi1, Yoshinobu Uno1, Chizuko Nishida2, Yoichi Matsuda1,3* 1 Laboratory of Animal Genetics, Department of Applied Molecular Biosciences, Graduate School of Bioagricultural Sciences, Nagoya University, Nagoya, Japan, 2 Department of Natural History Sciences, Faculty of Science, Hokkaido University, Sapporo, Japan, 3 Avian Bioscience Research Center, Graduate School of Bioagricultural Sciences, Nagoya University, Nagoya, Japan Abstract Reptiles have a wide diversity of sex-determining mechanisms and types of sex chromosomes. Turtles exhibit temperature- dependent sex determination and genotypic sex determination, with male heterogametic (XX/XY) and female heterogametic (ZZ/ZW) sex chromosomes. Identification of sex chromosomes in many turtle species and their comparative genomic analysis are of great significance to understand the evolutionary processes of sex determination and sex chromosome differentiation in Testudines. The Mexican giant musk turtle (Staurotypus triporcatus, Kinosternidae, Testudines) and the giant musk turtle (Staurotypus salvinii) have heteromorphic XY sex chromosomes with a low degree of morphological differentiation; however, their origin and linkage group are still unknown. Cross-species chromosome painting with chromosome-specific DNA from Chinese soft-shelled turtle (Pelodiscus sinensis) revealed that the X and Y chromosomes of S. triporcatus have homology with P. sinensis chromosome 6, which corresponds to the chicken Z chromosome. We cloned cDNA fragments of S. triporcatus homologs of 16 chicken Z-linked genes and mapped them to S. triporcatus and S. salvinii chromosomes using fluorescence in situ hybridization. Sixteen genes were localized to the X and Y long arms in the same order in both species. -

Sacred Turtles in Mayan Art and Iconography
Sacred Animals and Exotic Tropical Plants by Dr. Nicholas M. Hellmuth Sacred turtles in Mayan art and iconography new FLAAR Report* now lists all picted in Mayan art as one of the housings of the animals that were sacred or of God N (also called Pauahtun). Normally Aotherwise considered as special by God N is in a conch shell or snail shell, but the Classic Maya. !ere are animals that are sometimes he can be in a turtle carapace. related to the sky (constellations, stars, plan- Turtles are found in the various Mayan ets), the forests and those that are associated codices and in the murals of Bonampak. with rivers, lakes, swamps and the oceans. Any good book on Mayan archaeoastrono- !ese waters are conflated by the cosmology my will discuss the turtles (and peccary) in of the Preclassic and Classic Maya into the the murals and codices. Turtles are decora- underwaterworld. tion for a typical Puuc structure at Uxmal On the surface of this underwaterworld you in the Yucatán. Turtle symbolism is deeply get the major interaction of exotic creatures. embedded in Classic Maya beliefs. Crocodiles, fish, sharks, turtles—both sea tur- Dr. Nicholas M. Hellmuth is director of FLAAR tles and freshwater turtles—are often depicted. Reports (Foundation for Latin American Anthro- It has long been recognized that turtle pological Research). To view the list of sacred ani- mals visit www.maya-archaeology.org or contact carapaces were used as musical instruments. Dr. Hellmuth at frontdesk@flaar.org !ese instruments are pictured in murals * See the complete list of sacred animals at and on pottery vases, especially in the Late www.maya-archaeology.org Classic period (AD 600-800).