Colorectal Carcinoma: a General Overview and Future Perspectives in Colorectal Cancer
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Piggybac Mutagenesis and Exome Sequencing Identify Genetic Driver
Noorani et al. Genome Biology (2020) 21:181 https://doi.org/10.1186/s13059-020-02092-2 RESEARCH Open Access PiggyBac mutagenesis and exome sequencing identify genetic driver landscapes and potential therapeutic targets of EGFR-mutant gliomas Imran Noorani1,2* , Jorge de la Rosa1†, Yoon Ha Choi1,3†, Alexander Strong1, Hannes Ponstingl1, M. S. Vijayabaskar1, Jusung Lee3, Eunmin Lee3, Angela Richard-Londt4, Mathias Friedrich1,5, Federica Furlanetto5, Rocio Fuente1, Ruby Banerjee1, Fengtang Yang1, Frances Law1, Colin Watts2,6, Roland Rad5, George Vassiliou1, Jong Kyoung Kim3, Thomas Santarius2, Sebastian Brandner4 and Allan Bradley1* * Correspondence: [email protected]; [email protected] Abstract †Jorge de la Rosa and Yoonha Choi contributed equally to this work. Background: Glioma is the most common intrinsic brain tumor and also occurs in 1The Wellcome Trust Sanger the spinal cord. Activating EGFR mutations are common in IDH1 wild-type gliomas. Institute, Wellcome Trust Genome However, the cooperative partners of EGFR driving gliomagenesis remain poorly Campus, Hinxton, Cambridgeshire CB10 1SA, UK understood. Full list of author information is Results: We explore EGFR-mutant glioma evolution in conditional mutant mice by available at the end of the article whole-exome sequencing, transposon mutagenesis forward genetic screening, and transcriptomics. We show mutant EGFR is sufficient to initiate gliomagenesis in vivo, both in the brain and spinal cord. We identify significantly recurrent somatic alterations in these gliomas including mutant EGFR amplifications and Sub1, Trp53, and Tead2 loss-of-function mutations. Comprehensive functional characterization of 96 gliomas by genome-wide piggyBac insertional mutagenesis in vivo identifies 281 known and novel EGFR-cooperating driver genes, including Cdkn2a, Nf1, Spred1, and Nav3. -

(Kiss-1): Essential Players in Suppressing Tumor Metastasis
DOI:http://dx.doi.org/10.7314/APJCP.2013.14.11.6215 Kisspeptins (KiSS-1) as Supressors of Cancer Metastasis MINI-REVIEW Kisspeptins (KiSS-1): Essential Players in Suppressing Tumor Metastasis Venugopal Vinod Prabhu, Kunnathur Murugesan Sakthivel, Chandrasekharan Guruvayoorappan* Abstract Kisspeptins (KPs) encoded by the KiSS-1 gene are C-terminally amidated peptide products, including KP- 10, KP-13, KP-14 and KP-54, which are endogenous agonists for the G-protein coupled receptor-54 (GPR54). Functional analyses have demonstrated fundamental roles of KiSS-1 in whole body homeostasis including sexual differentiation of brain, action on sex steroids and metabolic regulation of fertility essential for human puberty and maintenance of adult reproduction. In addition, intensive recent investigations have provided substantial evidence suggesting roles of Kisspeptin signalling via its receptor GPR54 in the suppression of metastasis with a variety of cancers. The present review highlights the latest studies regarding the role of Kisspeptins and the KiSS-1 gene in tumor progression and also suggests targeting the KiSS-1/GPR54 system may represent a novel therapeutic approach for cancers. Further investigations are essential to elucidate the complex pathways regulated by the Kisspeptins and how these pathways might be involved in the suppression of metastasis across a range of cancers. Keywords: Kisspeptins - GPR54 receptor - KiSS-1 gene - metastasis - cancer Asian Pac J Cancer Prev, 14 (11), 6215-6220 Introduction deaths (Stafford et al., 2008). There are around 20 different metastasis suppressor genes which brings a valuable Cancer is a class of disease characterized by out-of- mechanistic insight for guiding specific therapeutic control cell growth. -

Essential Role of Autophagy in Protecting Neonatal Haematopoietic Stem Cells from Oxidative Stress in a P62-Independent Manner
www.nature.com/scientificreports OPEN Essential role of autophagy in protecting neonatal haematopoietic stem cells from oxidative stress in a p62‑independent manner Naho Nomura1,7,10, Chiaki Ito1,10, Takako Ooshio1,8, Yuko Tadokoro1,2, Susumu Kohno3, Masaya Ueno1,2, Masahiko Kobayashi1,2, Atsuko Kasahara4, Yusuke Takase1,9, Kenta Kurayoshi1, Sha Si1,2, Chiaki Takahashi3, Masaaki Komatsu5, Toru Yanagawa6 & Atsushi Hirao1,2* Autophagy is a cellular degradation system contributing to homeostasis of tissue stem cells including haematopoietic stem cells (HSCs). It plays pleiotropic roles in HSC characteristics throughout life, but its stage‑specifc roles in HSC self‑renewal are unclear. To investigate the efects of Atg5 deletion on stage‑specifc HSC functions, we compared the repopulating capacity of HSCs in Atg5f/f;Vavi-cre mice from postnatal day (P) 0–7 weeks of age. Interestingly, Atg5 defciency led to no remarkable abnormality in the HSC self‑renewal capacity at P0, but signifcant defects at P7, followed by severe defects. Induction of Atg5 deletion at P5 by tamoxifen administration to Atg5f/f;Rosa26-Cre-ERT2 mice resulted in normal haematopoiesis, including the HSC population, until around 1 year, suggesting that Atg5 in the early neonatal period was critical for haematopoiesis in adults. Mitochondrial oxidative stress was increased by Atg5 loss in neonatal HSC/progenitor cells. Although p62 had accumulated in immature bone marrow cells of Atg5f/f;Vavi-cre mice, p62 deletion did not restore defective HSC functions, indicating that Atg5‑dependent haematopoietic regulation in the developmental period was independent of p62. This study proposes a critical role of autophagy in HSC protection against harsh environments in the early neonatal stage, which is essential for healthy long‑term haematopoiesis. -

De Novo Profiling of Long Non-Coding Rnas Involved
International Journal of Molecular Sciences Article De Novo Profiling of Long Non-Coding RNAs Involved in MC-LR-Induced Liver Injury in Whitefish: Discovery and Perspectives Maciej Florczyk * , Paweł Brzuzan and Maciej Wo´zny Department of Environmental Biotechnology, Faculty of Geoengineering, University of Warmia and Mazury in Olsztyn, ul. Słoneczna 45G, 10-709 Olsztyn, Poland; [email protected] (P.B.); [email protected] (M.W.) * Correspondence: maciej.fl[email protected] Abstract: Microcystin-LR (MC-LR) is a potent hepatotoxin for which a substantial gap in knowledge persists regarding the underlying molecular mechanisms of liver toxicity and injury. Although long non-coding RNAs (lncRNAs) have been extensively studied in model organisms, our knowledge concerning the role of lncRNAs in liver injury is limited. Given that lncRNAs show low levels of sequence conservation, their role becomes even more unclear in non-model organisms without an annotated genome, like whitefish (Coregonus lavaretus). The objective of this study was to discover and profile aberrantly expressed polyadenylated lncRNAs that are involved in MC-LR-induced liver injury in whitefish. Using RNA sequencing (RNA-Seq) data, we de novo assembled a high-quality whitefish liver transcriptome. This enabled us to find 94 differentially expressed (DE) putative evolutionary conserved lncRNAs, such as MALAT1, HOTTIP, HOTAIR or HULC, and 4429 DE putative novel whitefish lncRNAs, which differed from annotated protein-coding transcripts (PCTs) in terms of minimum free energy, guanine-cytosine (GC) base-pair content and length. Additionally, we identified DE non-coding transcripts that might be 30 autonomous untranslated regions (30UTRs) Citation: Florczyk, M.; Brzuzan, P.; Wo´zny, M. -

Regulators at Every Step—How Micrornas Drive Tumor Cell Invasiveness and Metastasis
cancers Review Regulators at Every Step—How microRNAs Drive Tumor Cell Invasiveness and Metastasis 1,2,3, 1,2, 1, Tomasz M. Grzywa y , Klaudia Klicka y and Paweł K. Włodarski * 1 Department of Methodology, Medical University of Warsaw, 02-091 Warsaw, Poland; [email protected] (T.M.G.); [email protected] (K.K.) 2 Doctoral School, Medical University of Warsaw, 02-091 Warsaw, Poland 3 Department of Immunology, Medical University of Warsaw, 02-097 Warsaw, Poland * Correspondence: [email protected] These authors contributed equally to this work. y Received: 19 November 2020; Accepted: 7 December 2020; Published: 10 December 2020 Simple Summary: Tumor cell invasiveness and metastasis are key processes in cancer progression and are composed of many steps. All of them are regulated by multiple microRNAs that either promote or suppress tumor progression. Multiple studies demonstrated that microRNAs target the mRNAs of multiple genes involved in the regulation of cell motility, local invasion, and metastatic niche formation. Thus, microRNAs are promising biomarkers and therapeutic targets in oncology. Abstract: Tumor cell invasiveness and metastasis are the main causes of mortality in cancer. Tumor progression is composed of many steps, including primary tumor growth, local invasion, intravasation, survival in the circulation, pre-metastatic niche formation, and metastasis. All these steps are strictly controlled by microRNAs (miRNAs), small non-coding RNA that regulate gene expression at the post-transcriptional level. miRNAs can act as oncomiRs that promote tumor cell invasion and metastasis or as tumor suppressor miRNAs that inhibit tumor progression. These miRNAs regulate the actin cytoskeleton, the expression of extracellular matrix (ECM) receptors including integrins and ECM-remodeling enzymes comprising matrix metalloproteinases (MMPs), and regulate epithelial–mesenchymal transition (EMT), hence modulating cell migration and invasiveness. -

Decoding the Roles of Long Noncoding Rnas in Hepatocellular Carcinoma
International Journal of Molecular Sciences Review Decoding the Roles of Long Noncoding RNAs in Hepatocellular Carcinoma Lok-Sze Wong and Chun-Ming Wong * The State Key Laboratory of Liver Research, Department of Pathology, Li-Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, China; [email protected] * Correspondence: [email protected] Abstract: Hepatocellular carcinoma (HCC) is one of the most prevalent malignancies worldwide. HCC is associated with several etiological factors, including HBV/HCV infections, cirrhosis, and fatty liver diseases. However, the molecular mechanism underlying HCC development remains largely elusive. The advent of high-throughput sequencing has unveiled an unprecedented discovery of a plethora of long noncoding RNAs (lncRNAs). Despite the lack of coding capacity, lncRNAs have key roles in gene regulation through interacting with various biomolecules. It is increasingly evident that the dysregulation of lncRNAs is inextricably linked to HCC cancer phenotypes, suggesting that lncRNAs are potential prognostic markers and therapeutic targets. In light of the emerging research in the study of the regulatory roles of lncRNAs in HCC, we discuss the association of lncRNAs with HCC. We link the biological processes influenced by lncRNAs to cancer hallmarks in HCC and describe the associated functional mechanisms. This review sheds light on future research directions, including the potential therapeutic applications of lncRNAs. Keywords: long non-coding RNA; hepatocellular carcinoma; cancer hallmarks; biomarkers; thera- peutic strategies; HBV/HCV infections Citation: Wong, L.-S.; Wong, C.-M. Decoding the Roles of Long Noncoding RNAs in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 1. Introduction 3137. https://doi.org/10.3390/ Hepatocellular carcinoma (HCC) is one of the most prevalent cancers worldwide. -

Mir-126-3P Contributes to Sorafenib Resistance in Hepatocellular Carcinoma Via Downregulating SPRED1
38 Original Article Page 1 of 14 miR-126-3p contributes to sorafenib resistance in hepatocellular carcinoma via downregulating SPRED1 Wenliang Tan1,2#, Zhirong Lin1,2#, Xianqing Chen3, Wenxin Li4, Sicong Zhu1,5, Yingcheng Wei1,2, Liyun Huo1,2, Yajin Chen1,2, Changzhen Shang1,2 1Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China; 2Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China; 3Department of Hepatobiliary Surgery, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 4Department of Cardiology, the Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China; 5Department of Surgical Intensive Care Unit, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China Contributions: (I) Conception and design: C Shang, Y Chen; (II) Administrative support: C Shang, Y Chen; (III) Provision of study materials or patients: W Tan, Z Lin; (IV) Collection and assembly of data: S Zhu, Y Wei, L Huo; (V) Data analysis and interpretation: X Chen, W Li; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Changzhen Shang; Yajin Chen. Department of Hepatobiliary Surgery, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou 510120, China. Email: [email protected]; [email protected]. Background: Sorafenib can prolong the survival of patients with advanced hepatocellular carcinoma (HCC). However, drug resistance remains the main obstacle to improving its efficiency. This study aimed to explore the likely molecular mechanism of sorafenib resistance. Methods: Differentially expressed microRNAs (miRNAs) related to sorafenib response were analyzed with the Limma package in R software. -

Diet, Nutrients, Phytochemicals, and Cancer Metastasis Suppressor Genes
Cancer Metastasis Rev DOI 10.1007/s10555-012-9369-5 Diet, nutrients, phytochemicals, and cancer metastasis suppressor genes Gary G. Meadows # Springer Science+Business Media, LLC (outside the USA) 2012 Abstract The major factor in the morbidity and mortality of EGF Epidermal growth factor cancer patients is metastasis. There exists a relative lack of EGFR Epidermal growth factor receptor specific therapeutic approaches to control metastasis, and EPA Eicosapentaenoic acid this is a fruitful area for investigation. A healthy diet and IFN Interferon lifestyle not only can inhibit tumorigenesis but also can have KAI1 Kangai 1 a major impact on cancer progression and survival. Many MALL Mal-like chemicals found in edible plants are known to inhibit meta- MAPK Mitogen activated protein kinase static progression of cancer. While the mechanisms under- MKK Mitogen-activated protein kinase kinase lying antimetastatic activity of some phytochemicals are miR Micro RNA being delineated, the impact of diet, dietary components, NDRG N-myc downstream-regulated gene and various phytochemicals on metastasis suppressor genes NM23 Nonmetastatic gene 23 is underexplored. Epigenetic regulation of metastasis sup- PDCD Programmed cell death pressor genes promises to be a potentially important mech- PEBP Phosphatidylethanolamine binding protein anism by which dietary components can impact cancer PTEN Phosphatase and tensin homolog metastasis since many dietary constituents are known to RASSF Ras-associated domain family modulate gene expression. The review addresses this area RECK Reversion-inducing-cysteine-rich protein with of research as well as the current state of knowledge regard- kazal motifs ing the impact of diet, dietary components, and phytochem- RhoGDI2 Rho GDP-dissociation inhibitor 2 icals on metastasis suppressor genes. -

Affinity Purification of NF1 Protein–Protein Interactors Identifies
G C A T T A C G G C A T genes Article Affinity Purification of NF1 Protein–Protein Interactors Identifies Keratins and Neurofibromin Itself as Binding Partners Rachel M. Carnes 1, Robert A. Kesterson 1 , Bruce R. Korf 1, James A. Mobley 2 and Deeann Wallis 1,* 1 Department of Genetics, The University of Alabama at Birmingham, Birmingham, AL 35294, USA 2 Department of Anesthesiology and Perioperative Medicine, The University of Alabama at Birmingham, Birmingham, AL 35294, USA * Correspondence: [email protected]; Tel.: +1-205-934-2794; Fax: +1-205-975-4418 Received: 8 July 2019; Accepted: 19 August 2019; Published: 28 August 2019 Abstract: Neurofibromatosis Type 1 (NF1) is caused by pathogenic variants in the NF1 gene encoding neurofibromin. Definition of NF1 protein–protein interactions (PPIs) has been difficult and lacks replication, making it challenging to define binding partners that modulate its function. We created a novel tandem affinity purification (TAP) tag cloned in frame to the 3’ end of the full-length murine Nf1 cDNA (mNf1). We show that this cDNA is functional and expresses neurofibromin, His-Tag, and can correct p-ERK/ERK ratios in NF1 null HEK293 cells. We used this affinity tag to purify binding partners with Strep-Tactin®XT beads and subsequently, identified them via mass spectrometry (MS). We found the tagged mNf1 can affinity purify human neurofibromin and vice versa, indicating that neurofibromin oligomerizes. We identify 21 additional proteins with high confidence of interaction with neurofibromin. After Metacore network analysis of these 21 proteins, eight appear within the same network, primarily keratins regulated by estrogen receptors. -
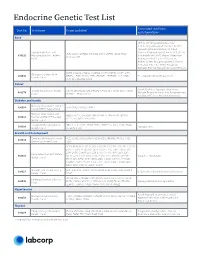
Endocrine Genetic Test List
Endocrine Genetic Test List Associated conditions Test No. Test Name Genes Included and phenotypes Bone HPP, X-Linked Hypophosphatemia, X-Linked Hypophosphatemic Rickets, XLH, Hypophosphatemic Rickets, X-Linked Hypophosphatasia and Dominant Hypophosphatemic Rickets (XLHR), ALPL, CLCN5, CYP2R1, CYP27B1, DMP1, ENPP1, FGF23, PHEX, 630292 Hypophosphatemic Rickets X-Linked Rickets (XLR), Vitamin D-Resistant SLC34A3, VDR Panel Rickets, X-Linked Vitamin D-Resistant Rickets (VDRR), Hypophosphatemic Vitamin D-Resistant Rickets (HPDR), Phosphate Diabetes, Familial Hypophosphatemic Rickets BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LRP5, Osteogenesis Imperfecta 630543 MBTPS2, P3H1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, SPARC, OI, Juvenile Primary Osteoporosis Genetic Panel TENT5A, TMEM38B, WNT1 Cancer Familial Isolated Hyperparathyroidism, VistaSeq® Endocrine Cancer CDC73, MAX, MEN1, NF1, PRKAR1A, PTEN, RET, SDHB, SDHC, SDHD, 481374 Multiple Endocrine Neoplasia, Paraganglioma, Panel* TMEM127, TP53 and VHL Parathyroid Cancer, Pheochromocytoma Diabetes and Insulin Maturity-Onset Diabetes of the 630568 GCK, HNF1A, HNF1B, HNF4A Young (MODY) 4-gene Panel Maturity-Onset Diabetes of ABCC8, APPL1, BLK, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, 630513 the Young (MODY) Expanded KLF11, NEUROD1, PAX4, PDX1 Genetic Panel Congenital Hyperinsulinism ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, KCNJ11, PGM1, PMM2, 630500 Hypoglycemia Genetic Panel SLC16A1, UCP2 Growth and Development Combined Pituitary Hormone GLI1, HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, -

Comprehensive Inherited Cancer Precision Panel Overview
Comprehensive Inherited Cancer Precision Panel Overview Hereditary cancer syndromes are encountered in all medical specialties. Although they account for about 5% of all malignancies, it is of special importance to identify these patients because, unlike patients with sporadic cancers, they require special, long-term care as their predisposition can cause them to develop certain tumors at a relatively early age. These cancers can arise in the lungs, kidneys, liver, pancreas, skin, eyes, heart. Most hereditary cancers are associated with a “germline mutation” that will be present in every cell of the human body. Identification of patients at risk of inherited cancer susceptibility is dependent upon the ability to characterize genes and alterations associated with increased cancer risk as well as gathering a detailed personal and family history aiding in the identification of the mode of inheritance as well as other family members at risk of suffering from this susceptibility. Most hereditary cancer syndromes follow an autosomal dominant inheritance, and the penetrance is high. The Igenomix Comprehensive Inherited Cancer Precision Panel provides a comprehensive analysis of the most common hereditary cancer syndromes using next-generation sequencing (NGS) to fully understand the spectrum of relevant cancer predisposition genes. Indications The Igenomix Comprehensive Inherited Caner Precision Panel is indicated as a screening and diagnostic test in those cases where there are: ‐ Multiple relatives on the same side of the family with the same or related forms of cancer ‐ Cancer at an early age ‐ Early presentation of an aggressive cancer type ‐ Multiple primary cancers in an individual 1 Clinical Utility The clinical utility of this panel is: ‐ Early and accurate genetic diagnosis allowing the most appropriate clinical management of a patient with personal or family history suggestive of a hereditary cancer syndrome. -

MRTFB Suppresses Colorectal Cancer Development Through Regulating SPDL1 and MCAM
MRTFB suppresses colorectal cancer development through regulating SPDL1 and MCAM Takahiro Kodamaa,b,c, Teresa A. Mariana,b, Hubert Leea, Michiko Kodamaa, Jian Lid, Michael S. Parmacekd, Nancy A. Jenkinsa,e, Neal G. Copelanda,e,1, and Zhubo Weia,b,1 aHouston Methodist Research Institute, Houston Methodist Hospital, Houston, TX 77030; bHouston Methodist Cancer Center, Houston Methodist Hospital, Houston, TX 77030; cDepartment of Gastroenterology and Hepatology, Graduate School of Medicine, Osaka University, 5650871 Suita, Osaka, Japan; dDepartment of Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104; and eGenetics Department, The University of Texas MD Anderson Cancer Center, Houston, TX 77030 Contributed by Neal G. Copeland, October 7, 2019 (sent for review June 18, 2019; reviewed by Masaki Mori and Hiroshi Seno) Myocardin-related transcription factor B (MRTFB) is a candidate tumor- shown to regulate cell cycle progression (9) and HCC xenograft suppressor gene identified in transposon mutagenesis screens of the tumor growth (10). Functional validation using cell culture sys- intestine, liver, and pancreas. Using a combination of cell-based assays, tems have also shown that reduced expression of MRTFB by in vivo tumor xenograft assays, and Mrtfb knockout mice, we demon- RNA interference leads to increased CRC cell invasion (4), strate here that MRTFB is a human and mouse colorectal cancer suggesting its important role in tumor progression. (CRC) tumor suppressor that functions in part by inhibiting cell Based on these results, we decided to conditionally delete invasion and migration. To identify possible MRTFB transcriptional Mrtfb in the mouse intestine to further explore its role in CRC.