Landscape of Tumor Suppressor Long Noncoding Rnas in Breast Cancer Boran Pang1, Qin Wang2, Shipeng Ning2, Junqiang Wu2, Xingda Zhang2, Yanbo Chen2 and Shouping Xu2*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hippo Signaling in Cancer: Lessons from Drosophila Models
fcell-07-00085 May 22, 2019 Time: 18:12 # 1 REVIEW published: 24 May 2019 doi: 10.3389/fcell.2019.00085 Hippo Signaling in Cancer: Lessons From Drosophila Models Kirti Snigdha1, Karishma Sanjay Gangwani1, Gauri Vijay Lapalikar2, Amit Singh1,3,4,5 and Madhuri Kango-Singh1,3,4,5* 1 Department of Biology, University of Dayton, Dayton, OH, United States, 2 Department of Biochemistry and Biophysics, Texas A&M University, College Station, TX, United States, 3 Pre-Medical Programs, University of Dayton, Dayton, OH, United States, 4 Center for Tissue Regeneration and Engineering at Dayton, University of Dayton, Dayton, OH, United States, 5 Integrated Science and Engineering Center, University of Dayton, Dayton, OH, United States Hippo pathway was initially identified through genetic screens for genes regulating organ size in fruitflies. Recent studies have highlighted the role of Hippo signaling as a key regulator of homeostasis, and in tumorigenesis. Hippo pathway is comprised of genes that act as tumor suppressor genes like hippo (hpo) and warts (wts), and oncogenes like yorkie (yki). YAP and TAZ are two related mammalian homologs of Drosophila Yki that act as effectors of the Hippo pathway. Hippo signaling deficiency can cause Edited by: YAP- or TAZ-dependent oncogene addiction for cancer cells. YAP and TAZ are often SrinivasVinod Saladi, activated in human malignant cancers. These transcriptional regulators may initiate Harvard Medical School, tumorigenic changes in solid tumors by inducing cancer stem cells and proliferation, United States culminating in metastasis and chemo-resistance. Given the complex mechanisms (e.g., Reviewed by: Sirisha M. Cheedipudi, of the cancer microenvironment, and the extrinsic and intrinsic cues) that overpower University of Texas Health Science YAP/TAZ inhibition, the molecular roles of the Hippo pathway in tumor growth and Center at Houston, United States Rizaldy Paz Scott, progression remain poorly defined. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Protein Identities in Evs Isolated from U87-MG GBM Cells As Determined by NG LC-MS/MS
Protein identities in EVs isolated from U87-MG GBM cells as determined by NG LC-MS/MS. No. Accession Description Σ Coverage Σ# Proteins Σ# Unique Peptides Σ# Peptides Σ# PSMs # AAs MW [kDa] calc. pI 1 A8MS94 Putative golgin subfamily A member 2-like protein 5 OS=Homo sapiens PE=5 SV=2 - [GG2L5_HUMAN] 100 1 1 7 88 110 12,03704523 5,681152344 2 P60660 Myosin light polypeptide 6 OS=Homo sapiens GN=MYL6 PE=1 SV=2 - [MYL6_HUMAN] 100 3 5 17 173 151 16,91913397 4,652832031 3 Q6ZYL4 General transcription factor IIH subunit 5 OS=Homo sapiens GN=GTF2H5 PE=1 SV=1 - [TF2H5_HUMAN] 98,59 1 1 4 13 71 8,048185945 4,652832031 4 P60709 Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 - [ACTB_HUMAN] 97,6 5 5 35 917 375 41,70973209 5,478027344 5 P13489 Ribonuclease inhibitor OS=Homo sapiens GN=RNH1 PE=1 SV=2 - [RINI_HUMAN] 96,75 1 12 37 173 461 49,94108966 4,817871094 6 P09382 Galectin-1 OS=Homo sapiens GN=LGALS1 PE=1 SV=2 - [LEG1_HUMAN] 96,3 1 7 14 283 135 14,70620005 5,503417969 7 P60174 Triosephosphate isomerase OS=Homo sapiens GN=TPI1 PE=1 SV=3 - [TPIS_HUMAN] 95,1 3 16 25 375 286 30,77169764 5,922363281 8 P04406 Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 - [G3P_HUMAN] 94,63 2 13 31 509 335 36,03039959 8,455566406 9 Q15185 Prostaglandin E synthase 3 OS=Homo sapiens GN=PTGES3 PE=1 SV=1 - [TEBP_HUMAN] 93,13 1 5 12 74 160 18,68541938 4,538574219 10 P09417 Dihydropteridine reductase OS=Homo sapiens GN=QDPR PE=1 SV=2 - [DHPR_HUMAN] 93,03 1 1 17 69 244 25,77302971 7,371582031 11 P01911 HLA class II histocompatibility antigen, -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Investigating Developmental and Functional Deficits in Neurodegenerative
UNIVERSITY OF CALIFORNIA, IRVINE Investigating developmental and functional deficits in neurodegenerative disease using transcriptomic analyses DISSERTATION submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences by Ryan Gar-Lok Lim Dissertation Committee: Professor Leslie M. Thompson, Chair Assistant Professor Dritan Agalliu Professor Peter Donovan Professor Suzanne Sandmeyer 2016 Introduction, Figure 1.1 © 2014 Macmillan Publishers Limited. Appendix 1 © 2016 Elsevier Ltd. All other materials © 2016 Ryan Gar-Lok Lim DEDICATION This dissertation is dedicated to my parents, sister, and my wife. I love you all very much and could not have accomplished any of this without your love and support. Please take the time to reflect back on all of the moments we’ve shared, and know, that it is because of those moments I have been able to succeed. This accomplishment is as much yours as it is mine. ii TABLE OF CONTENTS Page LIST OF FIGURES vi LIST OF TABLES ix ACKNOWLEDGMENTS x CURRICULUM VITAE xiii ABSTRACT OF THE DISSERTATION xv Introduction Huntington’s disease, the neurovascular unit and the blood-brain barrier 1 1.1 Huntington’s Disease 1.2 HTT structure and function 1.2.1 Normal HTT function and possible loss-of-function contributions to HD 1.3 mHTT pathogenesis 1.3.1 The dominant pathological features of mHTT - a gain-of- toxic function? 1.3.2 Cellular pathologies and non-neuronal contributions to HD 1.4 The neurovascular unit and the blood-brain barrier 1.4.1 Structure and function -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Epigenetic Mechanisms Are Involved in the Oncogenic Properties of ZNF518B in Colorectal Cancer
Epigenetic mechanisms are involved in the oncogenic properties of ZNF518B in colorectal cancer Francisco Gimeno-Valiente, Ángela L. Riffo-Campos, Luis Torres, Noelia Tarazona, Valentina Gambardella, Andrés Cervantes, Gerardo López-Rodas, Luis Franco and Josefa Castillo SUPPLEMENTARY METHODS 1. Selection of genomic sequences for ChIP analysis To select the sequences for ChIP analysis in the five putative target genes, namely, PADI3, ZDHHC2, RGS4, EFNA5 and KAT2B, the genomic region corresponding to the gene was downloaded from Ensembl. Then, zoom was applied to see in detail the promoter, enhancers and regulatory sequences. The details for HCT116 cells were then recovered and the target sequences for factor binding examined. Obviously, there are not data for ZNF518B, but special attention was paid to the target sequences of other zinc-finger containing factors. Finally, the regions that may putatively bind ZNF518B were selected and primers defining amplicons spanning such sequences were searched out. Supplementary Figure S3 gives the location of the amplicons used in each gene. 2. Obtaining the raw data and generating the BAM files for in silico analysis of the effects of EHMT2 and EZH2 silencing The data of siEZH2 (SRR6384524), siG9a (SRR6384526) and siNon-target (SRR6384521) in HCT116 cell line, were downloaded from SRA (Bioproject PRJNA422822, https://www.ncbi. nlm.nih.gov/bioproject/), using SRA-tolkit (https://ncbi.github.io/sra-tools/). All data correspond to RNAseq single end. doBasics = TRUE doAll = FALSE $ fastq-dump -I --split-files SRR6384524 Data quality was checked using the software fastqc (https://www.bioinformatics.babraham. ac.uk /projects/fastqc/). The first low quality removing nucleotides were removed using FASTX- Toolkit (http://hannonlab.cshl.edu/fastxtoolkit/). -

Genome-Wide Screening Identifies Genes and Biological Processes
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 10-12-2018 Genome-Wide Screening Identifies Genes and Biological Processes Implicated in Chemoresistance and Oncogene-Induced Apoptosis Tengyu Ko Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Cancer Biology Commons, Cell Biology Commons, and the Genomics Commons Recommended Citation Ko, Tengyu, "Genome-Wide Screening Identifies Genes and Biological Processes Implicated in Chemoresistance and Oncogene- Induced Apoptosis" (2018). LSU Doctoral Dissertations. 4715. https://digitalcommons.lsu.edu/gradschool_dissertations/4715 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. GENOME-WIDE SCREENING IDENTIFIES GENES AND BIOLOGICAL PROCESSES IMPLICATED IN CHEMORESISTANCE AND ONCOGENE- INDUCED APOPTOSIS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical and Veterinary Medical Sciences through the Department of Comparative Biomedical Sciences by Tengyu Ko B.S., University of California, Santa Barbara 2010 December 2018 ACKNOWLEDGEMENTS I would like to express my sincerest gratitude to my major supervisor Dr. Shisheng Li for giving me the opportunity to join his team and the freedom to pursue projects. I appreciate all of his thoughts and efforts. Truly, none of these findings would be possible without his supervisions, supports, insightful discussions, and patience. -

Review Article the HIPPO Pathway in Gynecological Malignancies
Am J Cancer Res 2020;10(2):610-629 www.ajcr.us /ISSN:2156-6976/ajcr0107590 Review Article The HIPPO pathway in gynecological malignancies Dongying Wang1, Jiaxing He1, Junxue Dong1,2, Thomas F Meyer2, Tianmin Xu1 1Department of Obstetrics and Gynecology, Second Hospital of Jilin University, Changchun, Jilin, P. R. China; 2De- partment of Molecular Biology, Max Planck Institute for Infection Biology, Berlin, Germany Received January 8, 2020; Accepted January 27, 2020; Epub February 1, 2020; Published February 15, 2020 Abstract: The Hippo pathway has been initially discovered by screening genes that regulate organ size in Drosophila. Recent studies have highlighted the role of the Hippo pathway in controlling organ size, tissue homeostasis and regeneration, and signaling dysregulation, especially the overactivation of the transcriptional coactivator YAP/TAZ, which leads to uncontrolled cell growth and malignant transformation. The core components of the Hippo pathway may initiate tumorigenesis by inducing tumor stem cells and proliferation, ultimately leading to metastasis and drug resistance, which occurs extensively in gynecological malignancies, including cervical cancer, ovarian cancer, and endometrial cancer. In this review, we attempt to systematically summarize recent progress in our understanding of the mechanism of Hippo pathway regulation in tumorigenesis and the mechanisms that underlie alterations during gynecological malignancies, as well as new therapeutic strategies. Keywords: Hippo pathway, YAP/TAZ, tumorigenesis, cervical cancer, ovarian cancer, endometrial cancer, thera- peutic strategies Background The Hippo pathway The Hippo pathway is a highly conserved signal- The Hippo pathway consists of a set of con- ing pathway in Drosophila and mammals that served kinases that can be divided into three controls organ size and tumor growth [1, 2]. -
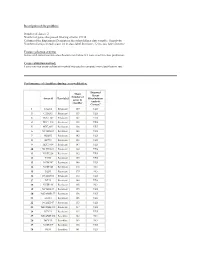
2 Number of Genes That Passed Filtering Criteria
Description of the problem: Number of classes: 2 Number of genes that passed filtering criteria: 25338 Column of the Experiment Descriptors sheet that defines class variable: Sensitivity Number of arrays in each class: 24 in class label Resistant , 32 in class label Sensitive Feature selection criteria: Genes with univariate mis-classfication rate below 0.2 were used for class prediction. Cross-validation method: Leave-one-out cross-validation method was used to compute mis-classification rate. Performance of classifiers during cross-validation. Diagonal Mean Linear Number of Array id Class label Discriminant genes in Analysis classifier Correct? 1 CALU1 Resistant 129 YES 2 CAMA1 Resistant 155 YES 3 HCC1143 Resistant 145 YES 4 HCC1419 Resistant 148 YES 5 HCC2935 Resistant 136 YES 6 NCIH2405 Resistant 148 YES 7 SKBR3 Resistant 142 YES 8 ZR751 Resistant 133 YES 9 HCC1954 Resistant 147 YES 10 NCIH1651 Resistant 139 YES 11 NCIH226 Resistant 142 YES 12 T47D Resistant 139 YES 13 NCIH747 Resistant 146 YES 14 NCIH345 Resistant 216 NO 15 DLD1 Resistant 179 NO 16 NCIH1666 Resistant 154 YES 17 BT20 Resistant 140 YES 18 NCIH196 Resistant 168 NO 19 NCIH1437 Resistant 159 YES 20 MDAMB157 Resistant 158 YES 21 LS123 Resistant 155 YES 22 NCIH2347 Resistant 133 YES 23 MDAMB361 Resistant 137 YES 24 BT474 Resistant 155 YES 25 MDAMB468 Sensitive 182 NO 26 MCF10 Sensitive 169 NO 27 NCIH209 Sensitive 128 YES 28 RKO Sensitive 141 YES 29 HCC70 Sensitive 182 NO 30 LS513 Sensitive 136 YES 31 NCIH1963 Sensitive 122 YES 32 HCC1187 Sensitive 145 YES 33 T84 Sensitive -

Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus–Host Protein Network
Published OnlineFirst September 12, 2018; DOI: 10.1158/2159-8290.CD-17-1018 RESEARCH ARTICLE Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus–Host Protein Network Manon Eckhardt1,2,3, Wei Zhang4, Andrew M. Gross4, John Von Dollen1,3, Jeffrey R. Johnson1,2,3, Kathleen E. Franks-Skiba1,3, Danielle L. Swaney1,2,3,5, Tasha L. Johnson3, Gwendolyn M. Jang1,3, Priya S. Shah1,3, Toni M. Brand6, Jacques Archambault7, Jason F. Kreisberg4,5, Jennifer R. Grandis5,6, Trey Ideker4,5, and Nevan J. Krogan1,2,3,5 ABSTRACT We have mapped a global network of virus–host protein interactions by purification of the complete set of human papillomavirus (HPV) proteins in multiple cell lines followed by mass spectrometry analysis. Integration of this map with tumor genome atlases shows that the virus targets human proteins frequently mutated in HPV− but not HPV+ cancers, providing a unique opportunity to identify novel oncogenic events phenocopied by HPV infection. For example, we find that the NRF2 transcriptional pathway, which protects against oxidative stress, is activated by interaction of the NRF2 regulator KEAP1 with the viral protein E1. We also demonstrate that the L2 HPV protein physically interacts with the RNF20/40 histone ubiquitination complex and promotes tumor cell inva- sion in an RNF20/40-dependent manner. This combined proteomic and genetic approach provides a systematic means to study the cellular mechanisms hijacked by virally induced cancers. SIGNIFICANCE: In this study, we created a protein–protein interaction network between HPV and human proteins. An integrative analysis of this network and 800 tumor mutation profiles identifies multiple oncogenesis pathways promoted by HPV interactions that phenocopy recurrent mutations in cancer, yielding an expanded definition of HPV oncogenic roles. -

Transcriptome Profiling Reveals the Complexity of Pirfenidone Effects in IPF
ERJ Express. Published on August 30, 2018 as doi: 10.1183/13993003.00564-2018 Early View Original article Transcriptome profiling reveals the complexity of pirfenidone effects in IPF Grazyna Kwapiszewska, Anna Gungl, Jochen Wilhelm, Leigh M. Marsh, Helene Thekkekara Puthenparampil, Katharina Sinn, Miroslava Didiasova, Walter Klepetko, Djuro Kosanovic, Ralph T. Schermuly, Lukasz Wujak, Benjamin Weiss, Liliana Schaefer, Marc Schneider, Michael Kreuter, Andrea Olschewski, Werner Seeger, Horst Olschewski, Malgorzata Wygrecka Please cite this article as: Kwapiszewska G, Gungl A, Wilhelm J, et al. Transcriptome profiling reveals the complexity of pirfenidone effects in IPF. Eur Respir J 2018; in press (https://doi.org/10.1183/13993003.00564-2018). This manuscript has recently been accepted for publication in the European Respiratory Journal. It is published here in its accepted form prior to copyediting and typesetting by our production team. After these production processes are complete and the authors have approved the resulting proofs, the article will move to the latest issue of the ERJ online. Copyright ©ERS 2018 Copyright 2018 by the European Respiratory Society. Transcriptome profiling reveals the complexity of pirfenidone effects in IPF Grazyna Kwapiszewska1,2, Anna Gungl2, Jochen Wilhelm3†, Leigh M. Marsh1, Helene Thekkekara Puthenparampil1, Katharina Sinn4, Miroslava Didiasova5, Walter Klepetko4, Djuro Kosanovic3, Ralph T. Schermuly3†, Lukasz Wujak5, Benjamin Weiss6, Liliana Schaefer7, Marc Schneider8†, Michael Kreuter8†, Andrea Olschewski1,