ADVANCES in N- and O-DEMETHYLATION of OPIATES
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Flavour Management by Citric Acid Negative MLF Starter Cultures Authors: Carsten Heinemeyer, 2B Fermcontrol; Ulrich Hamm DLR-RNH Bad Kreuznach, Germany; Dr
Flavour management by citric acid negative MLF starter cultures Authors: Carsten Heinemeyer, 2B FermControl; Ulrich Hamm DLR-RNH Bad Kreuznach, Germany; Dr. Jürgen Fröhlich, University of Mainz General will be completed in all fermentations. Additionally, acetic acid and a certain residual profile of the metabolic intermediates The malo-lactic fermentation (MLF) is a commonly used remain in the wine. method to convert the aggressive malic acid to lactic acid. This conversion results in a reduction of the titratable acidity, which New MLF Starter Cultures is desired mainly in red wine but also in numerous white wines. The development of a citric acid negative MLF starter culture This process will be done either by the indigenous flora of LAB gave a new opportunity to avoid diacetyl and acetic acid or by selected strains of LAB e.g. Oenococcus oeni. During the production from the citric acid degradation. The fermentation MLF Oenococcus oeni does not convert only malic acid into lactic with a citric acid negative MLF starter culture will best preserve acid, numerous amounts of aroma active by-products will also be the varietal character of the wine. produced. The best known is diacetyl, which gives buttery notes to the wine. Diacetyl will be produced during the MLF by the In a comprehensive four year study at the wine research institute conversion of the natural citric acid in the wine by Oenococcus DLR-RNH Bad Kreuznach in Rhineland-Palatinate-Germany, oeni (Jan Clair Nielsen 1999). Apart from the diacetyl formation different MLF starter strains were tested under practical by Oenococcus oeni, numerous intermediate by-products are winemaking conditions. -

An Original Method for Producing Acetaldehyde and Diacetyl by Yeast
b r a z i l i a n j o u r n a l o f m i c r o b i o l o g y 4 7 (2 0 1 6) 949–954 ht tp://www.bjmicrobiol.com.br/ Industrial Microbiology An original method for producing acetaldehyde and diacetyl by yeast fermentation a a,∗ a b a,c Irina Rosca , Anca Roxana Petrovici , Mihai Brebu , Irina Stoica , Bogdan Minea , a Narcisa Marangoci a “Petru Poni” Institute of Macromolecular Chemistry, Advanced Research Center for Bionanoconjugates and Biopolymers, Aleea GrigoreGhica Voda, Iasi, Romania b SC Zeelandia SRL, R&D Department, Valea Lupului, Iasi, Romania c “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania a r a t i c l e i n f o b s t r a c t Article history: In this study a natural culture medium that mimics the synthetic yeast peptone glucose Received 2 June 2015 medium used for yeast fermentations was designed to screen and select yeasts capable Accepted 29 February 2016 of producing high levels of diacetyl and acetaldehyde. The presence of whey powder and Available online 25 July 2016 sodium citrate in the medium along with manganese and magnesium sulfate enhanced Associate Editor: Jorge Gonzalo both biomass and aroma development. A total of 52 yeasts strains were cultivated in two Farias Avendano different culture media, namely, yeast peptone glucose medium and yeast acetaldehyde- diacetyl medium. The initial screening of the strains was based on the qualitative reaction Keywords: of the acetaldehyde with Schiff’s reagent (violet color) and diacetyl with Brady’s reagent Acetaldehyde (yellow precipitate). -

An Unrecognized Hazard in E-Cigarette Vapor: Preliminary Quantification of Methylglyoxal Formation from Propylene Glycol in E-Cigarettes
International Journal of Environmental Research and Public Health Article An Unrecognized Hazard in E-Cigarette Vapor: Preliminary Quantification of Methylglyoxal Formation from Propylene Glycol in E-Cigarettes Parham Azimi 1, Zahra Keshavarz 1, Marianne Lahaie Luna 1,2, Jose Guillermo Cedeno Laurent 1 , Jose Vallarino 1, David C. Christiani 1 and Joseph G. Allen 1,* 1 Department of Environmental Health, Harvard T. H. Chan School of Public Health, Boston, MA 02115, USA; [email protected] (P.A.); [email protected] (Z.K.); [email protected] (M.L.L.); [email protected] (J.G.C.L.); [email protected] (J.V.); [email protected] (D.C.C.) 2 Occupational & Environmental Health Division, Dalla Lana School of Public Health, University of Toronto, Toronto, ON M5T 3M7, Canada * Correspondence: [email protected] Abstract: Up to 95% of the liquid volume in an e-cigarette consists of propylene glycol. Previous research has shown that propylene glycol can generate diacetyl and formaldehyde when heated. New research shows that propylene glycol can also generate methylglyoxal, an alpha di-carbonyl compound recently shown to cause epithelial necrosis at even lower concentrations than diacetyl, the flavoring chemical associated with bronchiolitis obliterans (“Popcorn Lung”). We analyzed chemical emissions from 13 JUUL pod flavors. Diacetyl and methylglyoxal was detected in 100% of samples 3 3 with median concentration (range) of 20 µg/m (less than limit of quantification: 54 µg/m ) and 4219 µg/m3 (677–15,342 µg/m3), respectively. We also detected acetaldehyde (median concentration: Citation: Azimi, P.; Keshavarz, Z.; 341 µg/m3) and propionaldehyde (median concentration: 87 µg/m3) in all samples. -
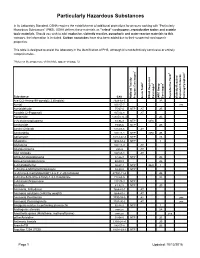
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

Recommendation from the Scientific Committee on Occupational Exposure Limits for Diacetyl
Employment, Social Affairs & Inclusion SCOEL Recommendation on Diacetyl Recommendation from the Scientific Committee on Occupational Exposure Limits for Diacetyl SCOEL/SUM/149 June 2014 June 2014 1 Employment, Social Affairs & Inclusion SCOEL Recommendation on Diacetyl Table of Contents 1. Substance identification, physico-chemical properties.............................................. 3 2. Occurrence/use and occupational exposure ........................................................... 3 2.1. Occurrence and use ...................................................................................... 3 2.2. Occupational exposure .................................................................................. 4 2.3. Methods of exposure monitoring and analysis .................................................. 5 3. Health significance ............................................................................................. 6 3.1. Toxicokinetics .............................................................................................. 7 3.1.1. Human data ........................................................................................... 7 3.1.2. Animal data ........................................................................................... 7 3.1.3. Biological monitoring ............................................................................... 8 3.2. Acute toxicity ............................................................................................... 8 3.2.1. Human data .......................................................................................... -

Substituted Deoxyguanosine Nucleosides from 2‐
Synthesis of N2-Substituted Deoxyguanosine UNIT 1.3 Nucleosides from 2-Fluoro-6-O- (Trimethylsilylethyl)-2′-Deoxyinosine This unit describes the synthesis of 2-fluoro-6-O-(trimethylsilylethyl)-2′-deoxyinosine and gives examples of its use for the preparation of N2-substituted deoxyguanosine nucleosides. Such nucleoside derivatives are used for a variety of purposes including chemotherapy, enzyme mechanism studies, nuclear magnetic resonance (NMR) studies (when isotopically labeled), and as synthetic standards for identification of adducts formed by the reaction of DNA with xenobiotics. In addition, the O6-protected 2-fluoro- 2′-deoxyinosine compounds can be converted to phosphoramidites and used in the synthesis of oligonucleotides, thus allowing substitution reactions to be carried out after oligonucleotide assembly. 2-Halopurine derivatives have been used for many years for the preparation of N2-sub- stituted guanosine derivatives, with the 2-fluoro substituent being the most easily dis- placed by nucleophiles (Montgomery and Hewson, 1960; Gerster and Robins, 1965, 1966). The 2-fluoro group is introduced by aqueous diazotization of guanosine in the presence of potassium fluoride or fluoroboric acid. However, these conditions are too harsh for 2′-deoxyguanosine and lead to depurination; hence, different synthetic method- ology is needed for the deoxynucleoside. The fluorine atom can be introduced success- fully by diazotization under anhydrous conditions with t-butyl nitrite as the diazotizing agent and HF in pyridine as the fluoride source (Robins and Uznanski, 1981; Lee et al., 1990; Harris et al., 1991). Success in the fluoridation step requires protection of the C6 oxygen group, which is done by Mitsunobu alkylation (Mitsunobu, 1981) with trimethyl- silylethanol or other alcohols. -

Diacetyl Take a Single Short Sniff
beer flavor standard 2,3-butanedione OFF-FLAVOR KIT available from www.cicerone.org ASSESSMENT CONFUSIONS Without covering the glass, swirl • Butyric acid the beer to release the aroma. • Vanillin Diacetyl Take a single short sniff. Repeat • Isobutyraldehyde “like butter, or as necessary. IMPORTANCE butter popcorn” THRESHOLD stouts 10 – 40 µg/l and lagers, eg in other lager beers. Considerable ORIGINS efforts are made by breweries to Produced in beer from a precursor formed by yeast during fermentation. Can also be formed REMARKS by contaminant lactic acid bacteria. 2,3-Butanedione is one of two vicinal diketones found in beer. O The ratio of diacetyl to CH3 pentanedione concentrations can H3 C be used as an indicator of bacterial contamination in beer. TION O A www.aroxa.com CAS NUMBER [email protected] 431-03-8 ©2013 CARA TECHNOLOGY LTD % POPUL RANDALLS ROAD LEATHERHEAD, SURREY FLAVOR THRESHOLD KT22 7RY, UK beer flavor standard dimethyl sulfide OFF-FLAVOR KIT available from www.cicerone.org ASSESSMENT CONFUSIONS Without covering the glass, swirl • Methyl thioacetate the beer to release the aroma. • Ethanethiol DMS Take a single short sniff. Repeat • Dimethyl trisulphide “like sweetcorn or as necessary. IMPORTANCE tomato sauce” THRESHOLD 30 - 50 µg/l beers. Excessive levels are indicative ORIGINS of growth of contaminant bacteria Formed from malt-derived during fermentation. precursors, primarily during wort production and – to a lesser extent – REMARKS during fermentation. The perception of dimethyl sulfide H3 C CH3 aromatic higher -

Diacetyl by Wine Making Lactic Acid Bacteria
Agric. Biol. Chem., 49 (7), 2147-2157, 1985 2147 Transformation of Citric Acid to Acetic Acid, Acetoin and Diacetyl by Wine Making Lactic Acid Bacteria Yoshimi Shimazu, Mikio Uehara and Masazumi Watanabe Food Research Laboratory, KikkomanCorporation, 399 Noda, Noda-shi, Chiba 278, Japan Received January 18, 1985 Adecrease in citric acid and increases in acetic acid, acetoin and diacetyl were found in the test red wine after inoculation of intact cells of Leuconostoc mesenteroides subsp. lactosum ATCC27307, a malo-lactic bacterium, grownon the malate plus citrate-medium. Citric acid in the buffer solution was transformed to acetic acid, acetoin and diacetyl in the pH range of 2 to 6 after inoculation with intact cells of this bacterial species. It was concluded that citric acid in wine making involving malo- lactic fermentation, at first, was converted by citrate lyase to acetic and oxaloacetic acids, and the latter was successively transformed by decarboxylation to pyruvic acid which was subsequently converted to acetoin, diacetyl and acetic acid. Both the activities of citrate lyase and acetoin formation from pyruvic acid in the dialyzed cell- free extract were optimal at pH 6.0. Divalent cations such as Mn2+ , Mg2+ , Co2 + and Zn2 + activated the citrate lyase. The citrate lyase was completely inhibited by EDTA,Hg2+ and Ag2 +. The acetoin formation from pyruvic acid was significantly stimulated by thiamine pyrophosphate and CoCl2, and inhibited by oxaloacetic acid. Specific activities of the citrate lyase and acetoin formation were considerably variable amongthe six strains of malo-lactic bacteria examined. Someactivities of irreversible reduction of diacetyl to acetoin were foundin the cell-free extracts of four of the malo- lactic bacteria strains and the optimal pH was 6.0 for this activity of Leu. -

Independent Toxicology Assessment for Diacetyl
Independent Toxicology Assessment for Diacetyl December 17, 2008 Andrew Maier, PhD, CIH, DABT Associate Director Toxicology Excellence for Risk Assessment Phone: 513-542-7475 x16 [email protected] Presentation Outline • Who is TERA? • What is TERA’s role? • Applying a systematic approach to health-based occupational limit development. • Some initial thoughts on diacetyl risk assessment. 2 Toxicology Excellence for Risk Assessment (TERA) Toxicology Excellence for Risk Assessment (TERA) is a non-profit, 501(c)(3) corporation organized for scientific and educational purposes. The mission of TERA is to inform the protection of public health by developing and communicating risk assessment values, improving risk assessment methods through research, and to educate on risk assessment issues. •Independent non-profit – funding from individual mission- related projects sponsored by government and industry •Objective approach with a focus on science •Scientific opinions are released to the public 3 Recent TERA Sponsors • Approximately 70% of TERA funding is from government. • U.S. EPA: - Support of IRIS Assessments - Science Support Document for NO2 NAAQS. - Exposure and effect progression for phosgene-induced fibrosis. • U.S. NIOSH: - Development of IDLH values. - Implementation of new skin notation methodology. • State of Texas: - Peer Review of ESL values (e.g., 1,3-butadiene). • Industry Consortia: - Chloropicrin acute inhalation values presented to U.S. and CA agencies. • Volunteer Groups: - Approximately 7% of TERA budget, including service on AIHA WEEL Committee. 4 TERA’s Role for Diacetyl Issue • TERA is being funded for this work by a consortium of food producers. • Provide an independent toxicology review document for diacetyl. • Ensure the document is made available to interested OEL-setting organizations and agencies as an aid to their deliberations. -

ALDC Prevents Diacetyl
P.O. BOX 593 • NORTHAMPTON, MA 01061 413 404 4830 MURPHYANDSON.COM ALDC prevents diacetyl. By breaking down alpha acetolactate during fermentation, ALDC inhibits production of off flavors, ensuring quality and increasing capacity. In commercial PRODUCT INFO brewery trials, ALDC from ALDC from Murphy & Son is added at the Murphy & Son time of yeast pitching an prevents the reduced the level formation of diacetyl fermentation. of diacetyl in finished beer As a result, diacetyl can be kept below the below the flavor threshold flavor threshold, ensuring the quality, flavor, compared to the and consistency of every brew. Without previous batch the need for long maturation, tank space is brewed with increased. ALDC. BENEFITS DRY HOP CREEP Is your dry hopping kicking up diacetyl? Keep the Reduce diacetyl production level below threshold by adding ALDC to the cooled Meet peak capacity demands wort at yeast pitching. Utilize vessels more efficiently DIACETYL BOMBS? Ensure packaged beer quality Prevent the release of diacetyl forming in packages by removing the precursor—alpha acetolactate— from each and every batch before packaging. APPLICATION How much to add 1–5 grams per barrel When to use Add to fermenter at time of yeast pitching Activity range pH: 4.0–7.0 temperature: 50–104°F HOW DOES IT WORK? ALDC is an enzyme that effectively bypass- STORAGE es the production of diacetyl during fermen- Temperature tation to make acetoin, a normal fermenta- 32-50°F tion byproduct, which has very little flavor. Do not allow to freeze. If this is your first time using ALDC, then Location Cool, sealed, and away from sunlight add a higher dose, i.e. -

United States Patent (19) 11) 4,450,277 Graf Et Al
United States Patent (19) 11) 4,450,277 Graf et al. 45 May 22, 1984 54 PREPARATION OF1-SUBSTITUTED 56 References Cited MIDAZOLES U.S. PATENT DOCUMENTS T5) Inventors: Fritz Graf, Speyer; Leopold Hupfer, 3,037,028 5/1962 Green .................................. 54.8/335 Friedelsheim, both of Fed. Rep. of Germany OTHER PUBLICATIONS Preliminary Brochure of BASF AG on 1- and 2-Me 73) Assignee: BASF Aktiengesellschaft, Fed. Rep. thylimidazole and 1,2-Dimethylimidazole Dated of Germany 9/1967. Lions, F., et al., J. Proc. Royal Soc. N.S.W., 74 (1941), 21 Appl. No.: 190,901 pp. 365-372. (22 Filed: Sep. 25, 1980 Primary Examiner-Richard A. Schwartz Attorney, Agent, or Firm-Keil & Witherspoon (30) Foreign Application Priority Data 57 ABSTRACT Oct. 8, 1979 IDE Fed. Rep. of Germany ....... 2940709 A process for the preparation of 1-substituted imidaz 51) Int. Cl. .................. C07D 233/58; C07D 233/60; oles by reacting an a-dicarbonyl compound with am CO7D 233/61 monia, an aldehyde and a primary amine in an aqueous 52 U.S. C. .................................... 548/346; 548/335; medium, in a single stage, at 20-150° C. 548/341; 548/342 58 Field of Search ................ 548/335, 346, 341, 342 3 Claims, No Drawings 4,450,277 ; 2 1 hyde, acetaldehyde, propionaldehyde, isobutyralde PREPARATION OF1-SUBSTITUTED hyde and benzaldehyde. MDAZOLES Examples of primary amines to be used as starting . components are monoamines of the formula X-NH2, The present invention relates to a novel process for 5 of the aliphatic, cycloaliphatic or aromatic series. Suit the preparation of 1-substituted imidazoles. -

Acetoin Diacetyl
Acetoin Diacetyl Method no.: 1012 Control no.: T-1012-FV-01-0811-M Target concentration: 0.05 ppm (TWA) (0.18 mg/m3) acetoin 0.05 ppm (TWA) (0.18 mg/m3) diacetyl OSHA PEL: none acetoin none diacetyl ACGIH TLV: none acetoin none diacetyl Procedure: Samples are collected by drawing workplace air through two tubes containing specially cleaned and dried silica gel connected in series. Samples are extracted and derivatized with a solution of 95:5 ethyl alcohol:water containing 2 mg/mL of O-(2, 3, 4, 5, 6-pentafluorobenzyl) hydroxylamine hydrochloride (PFBHA) and analyzed by gas chromatography using an electron capture detector (GC-ECD). Recommended sampling time 180 min at 0.05 L/min (9.0 L) (TWA) and sampling rate: 15 min at 0.2 L/min (3 L) (short term) Reliable quantitation limit: 1.49 ppb (5.37 μg/m3) acetoin 1.30 ppb (4.57 μg/m3) diacetyl Standard error of 5.06% acetoin estimate at the target 5.11% diacetyl concentration: Special requirements: Protect samplers from the light during and after sampling with aluminum foil or opaque tape. Status of method: Evaluated method. This method has been subjected to the established evaluation procedures of the OSHA Salt Lake Technical Center Methods Development Team. November 2008 Mary E. Eide Methods Development Team Industrial Hygiene Chemistry Division OSHA Salt Lake Technical Center Sandy UT 84070-6406 1 of 34 T-1012-FV-01-0811-M 1. General Discussion For assistance with accessibility problems in using figures and illustrations presented in this method, please contact Salt Lake Technical Center (SLTC) at (801) 233-4900.