Investigations of Olfactory Stem Cells As a Model for Schizophrenia
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2010 Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis Ryan Fassnacht Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Physiology Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/2246 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. Ryan C. Fassnacht 2010 All Rights Reserved Molecular Mechanisms Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University. by Ryan Christopher Fassnacht, B.S. Hampden Sydney University, 2005 M.S. Virginia Commonwealth University, 2010 Director: Valeria Mas, Ph.D., Associate Professor of Surgery and Pathology Division of Transplant Department of Surgery Virginia Commonwealth University Richmond, Virginia July 9, 2010 Acknowledgement The Author wishes to thank his family and close friends for their support. He would also like to thank the members of the molecular transplant team for their help and advice. This project would not have been possible with out the help of Dr. Valeria Mas and her endearing -

Neutral Protease (Dispase™ Equivalent Enzyme)
A cost-effective, improved preparation of purified Paenbacillus polymyxa neutral protease (Dispase™ equivalent enzyme) RC McCarthy, FE Dwulet, AG Breite, M Green, VitaCyte LLC, Indianapolis, Indiana, USA Background Results Dispase™ (trademark of Godo-Sushei) is a P. polymyxa neutral Crude PPNP was obtained from Gibco (Dispase II) or One explanation for the results reported on the left is the protease (PPNP) initially manufactured as a enzyme to replace trypsin Worthington Biochemicals (NPRO2). Purified PPNP was different purities of the enzymes. This is an acceptable for isolating cells from tissue or recovering adherent cells from tissue obtained from Sigma (Dispase I) or Worthington explanation for the differences in the crude and purified culture vessels. For many cells lines, PPNP was as effective or Biochemicals (NPRO). A comparison of the specific forms of Dispase and NPRO enzymes. However, further superior to trypsin in recovering adherent cells after in vitro culture. activities of these enzymes is shown below. As expected, analysis of those purified enzymes with BP Protease However, PPNP also has a unique characteristic for removing sheets of the crude enzymes have a specific activity about 10% of showed that a different molecular forms of PPNP detected epithelial cells from culture vessels. This lead to the increased use of the specific activities found with the purified NPRO and by analytical anion exchange chromatography likely this enzyme to improve understanding of the basic biology of skin and Dispase I. By contrast, the BP Protease has a specific accounted for this difference. BPNP Lot 140530 Desalted Run 140612 05001:1_UV BPNP Lot 140530 Desalted Run 140612 05001:1_Inject BPNP Lot 140530 Desalted Run 140612 05001:1_Logbook as an enzyme to prepare cells or biomaterials for therapeutic use. -

Neutral Protease (Dispase®) Product Highlights
BSE BSE cover icon NEUTRAL PROTEASE (DISPASE®) Primary Cell Isolation/Tissue Dissociation BSE Tissue Dissociation cover icon BSE simplied icon for use nearNeutral interior text protease (Dispase®) is a non-mammalian animal origin free (AOF) metallo, neutral protease, purified Can also be used as a “key” in Table ofby Contents, methodsetc. developed at Worthington. Its mild proteolytic action makes the enzyme especially suitable for the preparation of primary cells and secondary (subcultivation) in cell culture since it is gentle on cell DISSOC UE IAT membranes. This protease is also used as a secondary enzyme in cell isolation and tissue dissociation S IO IS N T BSE applications, commonly used with collagenase. Tissue Dissociation simplied icon for use near interior text Description Activity Code Cat. No. Size Price Can also be used as a “key” in Table of Contents DISSOC UE IAT Neutral Protease (Dispase®), Purified ≥ 4 Units LS02100 10 mg $ 68.00 S IO NPRO IS N T Animal Origin Free. Chromatographically per mg LS02104 50 mg 275.00 purified. A lyophilized powder. dry weight LS02108 Bulk Inquire Store at 2-8°C. Neutral Protease, Partially Purified ≥ 0.1 Units NPRO2 LS02109 1 gm $ 136.00 Animal Origin Free. Partially purified. per mg LS02111 5 gm 605.00 dry weight LS02112 Bulk Inquire A lyophilized powder. Store at 2-8°C. Characteristics of Neutral Protease (Dispase®) from Bacillus polymyxa: Molecular Weight: 36 kda. pH Optimum: Stable over a wide pH range: 4.0-9.0, optimum pH 5.9-7.0. Stability/Storage: Stable at 2-8°C for 12 months. -

Endopeptidase-24.11 in Pig Lymph Nodes Purification and Immunocytochemical Localization in Reticular Cells
Biochem. J. (1986) 236, 801-810 (Printed in Great Britain) 801 Endopeptidase-24.11 in pig lymph nodes Purification and immunocytochemical localization in reticular cells Michael A. BOWES and A. John KENNY M.R.C. Membrane Peptidase Research Group, Department of Biochemistry, University of Leeds, Leeds LS2 9JT, U.K. Endopeptidase-24.11 (EC 3.4.24.11), a widely distributed cell-surface endopeptidase in pig tissues, was purified by immunoaffinity chromatography from its second most abundant source, lymph nodes. The detergent-solubilized enzyme is a glycoprotein with an apparent subunit Mr of 91 000, by electrophoresis in the presence of SDS. This value is intermediate between those observed in preparations from kidney and intestine. The specific activity (1251-labelled insulin B-chain as substrate) was similar to that prepared from other sources. Immuno-peroxidase and immunofluorescent cytochemical methods with either a monoclonal antibody, GK7C2, or an affinity-purified polyclonal antiserum, RP109, were used to establish the distribution and localization of the antigen in lymph nodes. Examination of many nodes confirmed the variability of endopeptidase-24.11 content from node to node. Pig lymph nodes are composed of functionally discrete nodelets and are anatomically inverted, with medulla being located peripheral to the cortex. Endopeptidase- 24.11 was present in medulla, paracortex and cortex. The medulla, containing relatively few lymphocytes, stained more intensely than other zones. Lymphocyte-rich areas stained only weakly, but antigen was detectable in the centres of follicles and more strongly in a band surrounding them. The pattern of staining was reticular in appearance in all zones. In primary cell cultures, set up after enzymic disruption of nodes, the immuno-positive cells were found to be adherent to glass or plastic and to exhibit a fibroblastic morphology. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

(12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown Et Al
US 20030082511A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown et al. (43) Pub. Date: May 1, 2003 (54) IDENTIFICATION OF MODULATORY Publication Classification MOLECULES USING INDUCIBLE PROMOTERS (51) Int. Cl." ............................... C12O 1/00; C12O 1/68 (52) U.S. Cl. ..................................................... 435/4; 435/6 (76) Inventors: Steven J. Brown, San Diego, CA (US); Damien J. Dunnington, San Diego, CA (US); Imran Clark, San Diego, CA (57) ABSTRACT (US) Correspondence Address: Methods for identifying an ion channel modulator, a target David B. Waller & Associates membrane receptor modulator molecule, and other modula 5677 Oberlin Drive tory molecules are disclosed, as well as cells and vectors for Suit 214 use in those methods. A polynucleotide encoding target is San Diego, CA 92121 (US) provided in a cell under control of an inducible promoter, and candidate modulatory molecules are contacted with the (21) Appl. No.: 09/965,201 cell after induction of the promoter to ascertain whether a change in a measurable physiological parameter occurs as a (22) Filed: Sep. 25, 2001 result of the candidate modulatory molecule. Patent Application Publication May 1, 2003 Sheet 1 of 8 US 2003/0082511 A1 KCNC1 cDNA F.G. 1 Patent Application Publication May 1, 2003 Sheet 2 of 8 US 2003/0082511 A1 49 - -9 G C EH H EH N t R M h so as se W M M MP N FIG.2 Patent Application Publication May 1, 2003 Sheet 3 of 8 US 2003/0082511 A1 FG. 3 Patent Application Publication May 1, 2003 Sheet 4 of 8 US 2003/0082511 A1 KCNC1 ITREXCHO KC 150 mM KC 2000000 so 100 mM induced Uninduced Steady state O 100 200 300 400 500 600 700 Time (seconds) FIG. -

Supplemental Figure S1 Differentially Methylated Regions (Dmrs
Supplemental Figure S1 '$$#0#,2'**7+#2&7*2#"0#%'-,11 #25##,"'1#1#122#1 '!2-0'*"#.'!2'-,-$122,1'2'-,$0-+2- !"Q !"2-$%," $ 31',% 25-$-*" !&,%# ," ' 0RTRW 1 !32V-$$ !0'2#0'T - #.0#1#,22'-, -$ "'$$#0#,2'**7+#2&7*2#"%#,#11',.0#,2#1,"2&#'0 #&4'-022,1'2'-, #25##,"'$$#0#,2"'1#1#122#1T-*!)00-51',"'!2#&7.#0+#2&7*2#"%#,#1Q%0700-51 &7.-+#2&7*2#"%#,#1Q31',%25-$-*"!&,%#,"'0RTRW1!32V-$$!0'2#0'T-%#,#1 +#22&# -4#!0'2#0'22,1'2'-,$0-+$%2-$Q5#2�#$-0#*1-',!*3"#" %#,#15'2&V4*3#0RTRWT$$#!2#"%#,10#&'%&*'%&2#" 712#0'1)1#T Supplemental Figure S2 Validation of results from the HELP assay using Epityper MassarrayT #13*21 $0-+ 2&# 1$ 117 5#0# !-00#*2#" 5'2& /3,2'22'4# +#2&7*2'-, ,*78#" 7 '13*$'2#11007$-04V-,"6U-%#,#.0-+-2#00#%'-,1T11007 51.#0$-0+#"31',%**4'* *#1+.*#1T S Supplemental Fig. S1 A unique hypermethylated genes (methylation sites) 454 (481) 5693 (6747) 120 (122) NLMGUS NEWMM REL 2963 (3207) 1338 (1560) 5 (5) unique hypomethylated genes (methylation sites) B NEWMM 0 (0) MGUS 454 (481) 0 (0) NEWMM REL NL 3* (2) 2472 (3066) NEWMM 2963 REL (3207) 2* (2) MGUS 0 (0) REL 2 (2) NEWMM 0 (0) REL Supplemental Fig. S2 A B ARID4B DNMT3A Methylation by MassArray Methylation by MassArray 0 0.2 0.4 0.6 0.8 1 1.2 0.5 0.6 0.7 0.8 0.9 1 2 0 NL PC MGUS 1.5 -0.5 NEW MM 1 REL MM -1 0.5 -1.5 0 -2 -0.5 -1 -2.5 -1.5 -3 Methylation by HELP Assay Methylation by HELP Methylation by HELP Assay Methylation by HELP -2 -3.5 -2.5 -4 Supplemental tables "3..*#+#,2*6 *#"SS 9*','!*!&0!2#0'12'!1-$.2'#,21+.*#1 DZ_STAGE Age Gender Ethnicity MM isotype PCLI Cytogenetics -

Identification of Biomarkers of Metastatic Disease in Uveal
Identification of biomarkers of metastatic disease in uveal melanoma using proteomic analyses Thesis submitted in accordance with the requirements of the University of Liverpool for the degree of Doctor in Philosophy Martina Angi June 2015 To Mario, the wind beneath my wings 2 Acknowledgments First and foremost, I would like to acknowledge my primary supervisor, Prof. Sarah Coupland, for encouraging me to undergo a PhD and for supporting me in this long journey. I am truly grateful to Dr Helen Kalirai for being the person I could always turn to, for a word of advice on cell culture as much as on parenting skills. I would also like to acknowledge Prof. Bertil Damato for being an inspiration and a mentor; and Dr Sarah Lake and Dr Joseph Slupsky for their precious advice. I would like to thank Dawn, Haleh, Fidan and Fatima for becoming my family away from home, and the other members of the LOORG for the fruitful discussions and lovely cakes. I would like to acknowledge Prof. Heinrich Heimann and the clinical team at LOOC, especially Sisters Hebbar, Johnston, Hachuela and Kaye, for their admirable dedication to UM patients and for their invaluable support to clinical research. I would also like to thank the members of staff in St Paul’s theatre and Simon Biddolph and Anna Ikin in Pathology for their precious help in sample collection. I am grateful to Dr Rosalind Jenkins who guided my first steps in the mysterious word of proteomics, and to Dr Deb Simpsons and Prof. Rob Beynon for showing me its beauty. -
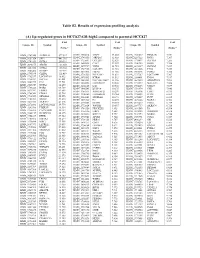
(A) Up-Regulated Genes in HCC827-GR-High2 Compared to Parental HCC827
Table S2. Results of expression profiling analysis (A) Up-regulated genes in HCC827-GR-high2 compared to parental HCC827 Fold Fold Fold Unique ID Symbol Unique ID Symbol Unique ID Symbol change* change* change* ILMN_1709348 ALDH1A1 577.587 ILMN_2310814 MAPT 13.003 ILMN_1741017 PIP4K2B 7.331 ILMN_1651354 SPP1 441.316 ILMN_1748650 MRPL45 12.988 ILMN_3237623 RNY1 7.297 ILMN_1701831 GSTA1 260.591 ILMN_1755897 UGT2B7 12.629 ILMN_1734897 SLC4A4 7.285 ILMN_1658835 CAV2 12.357 ILMN_1746359 RERG 7.280 ILMN_2094875 ABCB1 183.050 ILMN_1678939 VNN2 11.935 ILMN_1671337 SLC2A5 7.257 ILMN_3251540 GSTA2 145.982 ILMN_1729905 GAL3ST1 11.910 ILMN_1691606 LYG2 7.254 ILMN_2062468 IGFBP7 127.721 ILMN_1672536 FBLN1 11.716 ILMN_1785646 PMP22 7.246 ILMN_1795190 CLDN2 111.439 ILMN_1796339 PLEKHA2 11.631 ILMN_1737387 LOC728441 7.207 ILMN_1782937 LOC647169 98.612 ILMN_1676563 HTRA1 11.592 ILMN_1684401 FMO1 7.117 ILMN_1754247 SLC3A1 81.001 ILMN_3263423 LOC100129027 11.346 ILMN_1687035 ADAMTSL4 7.098 ILMN_1662795 CA2 79.581 ILMN_1694898 LOC653857 10.906 ILMN_2153572 MAGEA3 7.086 ILMN_2168747 GSTA2 66.250 ILMN_2404625 LAT 10.560 ILMN_1784283 USH1C 7.079 ILMN_1764228 DAB2 64.709 ILMN_1666546 DUSP14 10.375 ILMN_1731374 CPE 7.046 ILMN_1675797 EPDR1 63.605 ILMN_1764571 ARHGAP23 10.299 ILMN_1765446 EMP3 6.933 ILMN_1708341 PDZK1 59.714 ILMN_3200140 LOC645638 10.284 ILMN_1754002 IL1F8 6.863 ILMN_1713529 SEMA6A 52.575 ILMN_3244343 SNORA21 10.171 ILMN_1878007 FUT9 6.835 ILMN_1708391 NR1H4 43.218 ILMN_1671489 PC 10.075 ILMN_1699208 NAP1L1 6.763 ILMN_2412336 AKR1C2 42.826 ILMN_2404688 -

Supplementary Table 3 Gene Microarray Analysis: PRL+E2 Vs
Supplementary Table 3 Gene microarray analysis: PRL+E2 vs. control ID1 Field1 ID Symbol Name M Fold P Value 69 15562 206115_at EGR3 early growth response 3 2,36 5,13 4,51E-06 56 41486 232231_at RUNX2 runt-related transcription factor 2 2,01 4,02 6,78E-07 41 36660 227404_s_at EGR1 early growth response 1 1,99 3,97 2,20E-04 396 54249 36711_at MAFF v-maf musculoaponeurotic fibrosarcoma oncogene homolog F 1,92 3,79 7,54E-04 (avian) 42 13670 204222_s_at GLIPR1 GLI pathogenesis-related 1 (glioma) 1,91 3,76 2,20E-04 65 11080 201631_s_at IER3 immediate early response 3 1,81 3,50 3,50E-06 101 36952 227697_at SOCS3 suppressor of cytokine signaling 3 1,76 3,38 4,71E-05 16 15514 206067_s_at WT1 Wilms tumor 1 1,74 3,34 1,87E-04 171 47873 238623_at NA NA 1,72 3,30 1,10E-04 600 14687 205239_at AREG amphiregulin (schwannoma-derived growth factor) 1,71 3,26 1,51E-03 256 36997 227742_at CLIC6 chloride intracellular channel 6 1,69 3,23 3,52E-04 14 15038 205590_at RASGRP1 RAS guanyl releasing protein 1 (calcium and DAG-regulated) 1,68 3,20 1,87E-04 55 33237 223961_s_at CISH cytokine inducible SH2-containing protein 1,67 3,19 6,49E-07 78 32152 222872_x_at OBFC2A oligonucleotide/oligosaccharide-binding fold containing 2A 1,66 3,15 1,23E-05 1969 32201 222921_s_at HEY2 hairy/enhancer-of-split related with YRPW motif 2 1,64 3,12 1,78E-02 122 13463 204015_s_at DUSP4 dual specificity phosphatase 4 1,61 3,06 5,97E-05 173 36466 227210_at NA NA 1,60 3,04 1,10E-04 117 40525 231270_at CA13 carbonic anhydrase XIII 1,59 3,02 5,62E-05 81 42339 233085_s_at OBFC2A oligonucleotide/oligosaccharide-binding