Characterizing Nav1.5 Expression, Organization, and Electrical Behavior in Cardiomyocyte Domains
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Autosomal Recessive Ataxias and with the Early-Onset Parkinson’S Disease That We Identified Here As Overlapping with the SFARI Autism Gene
Roadmap to Help Develop Personalized Targeted Treatments for Autism as a Disorder of the Nervous Systems Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) 1 | P a g e Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -
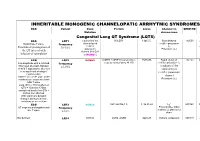
Inheritable Arrhytmia Syndromes
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT. -

Comparative Transcriptome Profiling of the Human and Mouse Dorsal Root Ganglia: an RNA-Seq-Based Resource for Pain and Sensory Neuroscience Research
bioRxiv preprint doi: https://doi.org/10.1101/165431; this version posted October 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq-based resource for pain and sensory neuroscience research Short Title: Human and mouse DRG comparative transcriptomics Pradipta Ray 1, 2 #, Andrew Torck 1 , Lilyana Quigley 1, Andi Wangzhou 1, Matthew Neiman 1, Chandranshu Rao 1, Tiffany Lam 1, Ji-Young Kim 1, Tae Hoon Kim 2, Michael Q. Zhang 2, Gregory Dussor 1 and Theodore J. Price 1, # 1 The University of Texas at Dallas, School of Behavioral and Brain Sciences 2 The University of Texas at Dallas, Department of Biological Sciences # Corresponding authors Theodore J Price Pradipta Ray School of Behavioral and Brain Sciences School of Behavioral and Brain Sciences The University of Texas at Dallas The University of Texas at Dallas BSB 14.102G BSB 10.608 800 W Campbell Rd 800 W Campbell Rd Richardson TX 75080 Richardson TX 75080 972-883-4311 972-883-7262 [email protected] [email protected] Number of pages: 27 Number of figures: 9 Number of tables: 8 Supplementary Figures: 4 Supplementary Files: 6 Word count: Abstract = 219; Introduction = 457; Discussion = 1094 Conflict of interest: The authors declare no conflicts of interest Patient anonymity and informed consent: Informed consent for human tissue sources were obtained by Anabios, Inc. (San Diego, CA). Human studies: This work was approved by The University of Texas at Dallas Institutional Review Board (MR 15-237). -

The ERK MAPK Pathway Modulates Gq-Dependent Locomotion In
bioRxiv preprint doi: https://doi.org/10.1101/231746; this version posted December 10, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 The ERK MAPK pathway modulates Gq-dependent locomotion in 2 Caenorhabditis elegans 3 4 5 Brantley Coleman, Irini Topalidou, and Michael Ailion 6 7 8 9 10 11 12 Department of Biochemistry, University of Washington, Seattle, WA, 98195 13 1 bioRxiv preprint doi: https://doi.org/10.1101/231746; this version posted December 10, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 14 Running Title: The ERK pathway modulates Gq signaling 15 16 Keywords: Gq signaling, ERK MAPK pathway, Rho small GTPase, C. elegans, G 17 protein 18 19 Corresponding author: 20 Michael Ailion 21 Department of Biochemistry 22 University of Washington 23 Box 357350 24 1705 NE Pacific St 25 Seattle, WA 98195 26 Phone: 206-685-0111 27 email: [email protected] 28 2 bioRxiv preprint doi: https://doi.org/10.1101/231746; this version posted December 10, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Sodium Channel Mutations in Epilepsy and Other Neurological Disorders
Sodium channel mutations in epilepsy and other neurological disorders Miriam H. Meisler, Jennifer A. Kearney J Clin Invest. 2005;115(8):2010-2017. https://doi.org/10.1172/JCI25466. Review Series Since the first mutations of the neuronal sodium channelS CN1A were identified 5 years ago, more than 150 mutations have been described in patients with epilepsy. Many are sporadic mutations and cause loss of function, which demonstrates haploinsufficiency of SCN1A. Mutations resulting in persistent sodium current are also common. Coding variants of SCN2A, SCN8A, and SCN9A have also been identified in patients with seizures, ataxia, and sensitivity to pain, respectively. The rapid pace of discoveries suggests that sodium channel mutations are significant factors in the etiology of neurological disease and may contribute to psychiatric disorders as well. Find the latest version: https://jci.me/25466/pdf Review series Sodium channel mutations in epilepsy and other neurological disorders Miriam H. Meisler and Jennifer A. Kearney Department of Human Genetics, University of Michigan, Ann Arbor, Michigan, USA. Since the first mutations of the neuronal sodium channel SCN1A were identified 5 years ago, more than 150 muta- tions have been described in patients with epilepsy. Many are sporadic mutations and cause loss of function, which demonstrates haploinsufficiency of SCN1A. Mutations resulting in persistent sodium current are also common. Coding variants of SCN2A, SCN8A, and SCN9A have also been identified in patients with seizures, ataxia, and sen- sitivity to pain, respectively. The rapid pace of discoveries suggests that sodium channel mutations are significant factors in the etiology of neurological disease and may contribute to psychiatric disorders as well. -

Molecular Mapping of the Binding Sites of a Nav1.1 Activator
Molecular Mapping of the Binding Sites of a Nav1.1 Activator Héctor Botella Master’s Thesis by Héctor Botella, MSc in MedIS Biomedicine, Aalborg Universitet / H. Lundbeck A/S (2018) Thesis submitted 07 - 09 - 2018 Internal Thesis supervisor Prof. Dr. Meg Duroux, Aalborg University External Thesis supervisor Dr. Tau Benned-Jensen, Lundbeck A/S Thesis committee Prof. Dr. Sebastian Frische Prof. Dr. Meg Duroux, Aalborg University Dr. Tau Benned-Jensen, Lundbeck A/S Thesis Series Aalborg University 9220 Aalborg Ø, Aalborg, Denmark Faculty of Medicine, Aalborg University Niels Jernes Vej 10 | DK - 9220 Aalborg Ø Phone: +45 9940 9940 | [email protected] School of Medicine and Health Niels Jernes Vej 12, A5 | 9220 Aalborg Ø | Aalborg, Denmark Department of Health, Science and Technology Aalborg University | Fredrik Bajers Vej 7 D2 | DK-9220, Aalborg Ø Published in Aalborg University Publication Repository (vbn.aau.dk) © Héctor J. Botella Valle Printed in Aalborg, Denmark, 2018 Nº of pages 95 2 Molecular Mapping of the Binding Sites of a Nav1.1 Activator Master’s Thesis by Héctor Botella, MSc in MedIS Biomedicine, Aalborg Universitet / H. Lundbeck A/S (2018) I. INDEX OF CONTENTS I. Index of contents ................................................................................................................................................................. 3 II. Preface ................................................................................................................................................................................ -

The SEK-1 P38 MAP Kinase Pathway Modulates Gq Signaling in Caenorhabditis Elegans
INVESTIGATION The SEK-1 p38 MAP Kinase Pathway Modulates Gq Signaling in Caenorhabditis elegans Jill M. Hoyt, Samuel K. Wilson, Madhuri Kasa, Jeremy S. Rise, Irini Topalidou, and Michael Ailion1 Department of Biochemistry, University of Washington, Seattle, Washington 98195 ORCID ID: 0000-0002-8070-1362 (M.A.) ABSTRACT Gq is a heterotrimeric G protein that is widely expressed in neurons and regulates neuronal KEYWORDS activity. To identify pathways regulating neuronal Gq signaling, we performed a forward genetic screen in p38 MAPK Caenorhabditis elegans for suppressors of activated Gq. One of the suppressors is an allele of sek-1, which pathway encodes a mitogen-activated protein kinase kinase (MAPKK) in the p38 MAPK pathway. Here, we show that Gq signaling sek-1 mutants have a slow locomotion rate and that sek-1 acts in acetylcholine neurons to modulate both animal behavior locomotion rate and Gq signaling. Furthermore, we find that sek-1 acts in mature neurons to modulate C. elegans locomotion. Using genetic and behavioral approaches, we demonstrate that other components of the p38 MAPK pathway also play a positive role in modulating locomotion and Gq signaling. Finally, we find that mutants in the SEK-1 p38 MAPK pathway partially suppress an activated mutant of the sodium leak channel, NCA-1/NALCN, a downstream target of Gq signaling. Our results suggest that the SEK-1 p38 pathway may modulate the output of Gq signaling through NCA-1(unc-77). Gq is a widely expressed heterotrimeric G protein that regulates a variety ing pathway (Koelle 2016). In neurons, EGL-30 signals through EGL-8 of biological processes, ranging from neurotransmission to cardiovas- (PLCb) (Lackner et al. -

A Biphasic Role for the Voltage-Gated Sodium Channel Scn5lab
A BIPHASIC ROLE FOR THE VOLTAGE-GATED SODIUM CHANNEL SCN5LAB IN CARDIAC DEVELOPMENT OF ZEBRAFISH By Jeffrey Scott Bennett Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Human Genetics August, 2013 Nashville, TN Approved: H. Scott Baldwin, M.D. Dan Roden, M.D. Al George, M.D. Doug Mortlock, Ph.D. Charles Hong, M.D., Ph.D. To those who came before me, on whose shoulders I stand, and Amanda, on whose shoulders I lean. ii ACKNOWLEDGEMENTS This work would not have been possible without the assistance and support of many people and departments throughout the Vanderbilt community. I’d like to particularly thank Terry Dermody, and the leaders and administration of the Medical Scientist Training Program for years of support and encouragement, as well as the Graduate Program in Human Genetics, for providing me an academic home for these years. So much of my academic success, present and future, is owed to the many people who got me to where I am today, and prepared me for tomorrow. My career never could have gotten started without the unfailing support of my mentor, Dan Roden. I am blessed to have been welcomed into a laboratory which truly values science and discovery, and given all I needed to be put in a position to succeed. I hope my future success reflects upon the training and commitment I received from him. Additionally, much of the assistance after I first joined lab, as well as my knowledge of developmental biology and the zebrafish model system comes from Dina Myers Stroud, who graciously served as a day‐ to‐day mentor, teaching skills and techniques I would use over the rest of my career. -

Androgen-Induced Programs for Prostate Epithelial Growth and Invasion Arise in Embryogenesis and Are Reactivated in Cancer
Oncogene (2008) 27, 7180–7191 & 2008 Macmillan Publishers Limited All rights reserved 0950-9232/08 $32.00 www.nature.com/onc ORIGINAL ARTICLE Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer EM Schaeffer1,2,3,5, L Marchionni3,5, Z Huang1,2, B Simons1, A Blackman3,WYu3, G Parmigiani1,3,4 and DM Berman1,2,3 1Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 2Department of Urology, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 3Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, MD, USA and 4Department of Biostatistics, Johns Hopkins University School of Medicine, Baltimore, MD, USA Cancer cells differentiate along specific lineages that most significant links to the development and cancer, we largelydetermine their clinical and biologic behavior. highlight coordinate induction of the transcription factor Distinct cancer phenotypes from different cells and organs Sox9 and suppression of the proapoptotic phospholipid- likelyresult from unique gene expression repertoires binding protein Annexin A1 that link earlyprostate established in the embryo and maintained after malignant development to earlyprostate carcinogenesis. These transformation. We used comprehensive gene expression results credential earlyprostate development as a reliable analysis to examine this concept in the prostate, an organ and valid model system for the investigation of genes and with a tractable developmental program and a high pathways that drive prostate cancer. propensityfor cancer. We focused on gene expression in Oncogene (2008) 27, 7180–7191; doi:10.1038/onc.2008.327; the murine prostate rudiment at three time points during published online 15 September 2008 the first 48 h of exposure to androgen, which initiates proliferation and invasion of prostate epithelial buds into Keywords: prostate organogenesis; androgen signaling; surrounding urogenital sinus mesenchyme. -

Kinds of Mutations Mutations at Single DNA Bases Chromosome (Karyotype) Mutations
Mutations at single DNA bases Kinds of mutations 1. Point mutations (change in a single DNA base) 2. Chromosomal mutations • Insertions & deletions • Inversions • Gene duplications • Polyploidizations Transitions are more frequent than transversions Synonymous (“silent”) mutations: Chromosome (karyotype) mutations Changes in the 3rd base of a codon that do not change the amino acid of the protein made by • Karyotype: The chromosomal arrangement of an that gene (because of redundancy in the individual (or species) genetic code) • Changes in the karyotype – Deletions and insertions – Inversions Non-synonymous (replacement) mutations: – Gene duplications – Polyploidization Changes in a DNA sequence that do change an amino acid 1 Duplication: Inversion: A doubling of a part of a chromosome, A reversal of the orientation of a of an entire chromosome, or even the part of a chromosome whole genome Tandem duplication resulting from unequal crossing-over during meiosis Synteny between human and mouse MGSC 2002 Human, Chimp, Gorilla, Orangutan: 2N=46 Mouse: 2N=40 Down’s Syndrome Male (21 triploidy) Rice: 2N=24 2 The standard model of genome evolution What is the origin of novelty, and diversity? DNA sequence The engine : Mutations Selection (purifying or positive) The steering wheel : Random drift of neutral mutations DNA sequence (Altered) Or, how do you acquire new function while maintaining the old? Carroll S.B. et al. From DNA to Diversity (2001) Blackwell Science Evolution by Gene Duplication Susumu Ohno, 1970 A new function is created by -

Association of Rare Missense Variants in the Second Intracellular Loop of Nav1.7 Sodium Channels with Familial Autism
OPEN Molecular Psychiatry (2018) 23, 231–239 www.nature.com/mp ORIGINAL ARTICLE Association of rare missense variants in the second intracellular loop of NaV1.7 sodium channels with familial autism M Rubinstein1,6,7, A Patowary2,7, IB Stanaway3, E McCord1, RR Nesbitt2, M Archer2, T Scheuer1, D Nickerson3, WH Raskind2,4, EM Wijsman3,4,5, R Bernier2, WA Catterall1 and Z Brkanac2 Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder often accompanied by intellectual disability, language impairment and medical co-morbidities. The heritability of autism is high and multiple genes have been implicated as causal. However, most of these genes have been identified in de novo cases. To further the understanding of familial autism, we performed whole-exome sequencing on five families in which second- and third-degree relatives were affected. By focusing on novel and protein-altering variants, we identified a small set of candidate genes. Among these, a novel private missense C1143F variant in the second intracellular loop of the voltage-gated sodium channel NaV1.7, encoded by the SCN9A gene, was identified in one family. C1143F Through electrophysiological analysis, we show that NaV1.7 exhibits partial loss-of-function effects, resulting in slower recovery from inactivation and decreased excitability in cultured cortical neurons. Furthermore, for the same intracellular loop of NaV1.7, we found an excess of rare variants in a case–control variant-burden study. Functional analysis of one of these variants, M932L/V991L, also demonstrated reduced firing in cortical neurons. However, although this variant is rare in Caucasians, it is frequent in Latino population, suggesting that genetic background can alter its effects on phenotype. -
Modulation of Gq-Rho Signaling by the ERK MAPK Pathway Controls Locomotion in Caenorhabditis Elegans
| INVESTIGATION Modulation of Gq-Rho Signaling by the ERK MAPK Pathway Controls Locomotion in Caenorhabditis elegans Brantley Coleman, Irini Topalidou, and Michael Ailion1 Department of Biochemistry, University of Washington, Seattle, Washington 98195 ORCID ID: 0000-0002-8070-1362 (M.A.) ABSTRACT The heterotrimeric G protein Gq regulates neuronal activity through distinct downstream effector pathways. In addition to the canonical Gq effector phospholipase Cb, the small GTPase Rho was recently identified as a conserved effector of Gq. To identify additional molecules important for Gq signaling in neurons, we performed a forward genetic screen in the nematode Caenorhabditis elegans for suppressors of the hyperactivity and exaggerated waveform of an activated Gq mutant. We isolated two mutations affecting the MAP kinase scaffold protein KSR-1 and found that KSR-1 modulates locomotion downstream of, or in parallel to, the Gq-Rho pathway. Through epistasis experiments, we found that the core ERK MAPK cascade is required for Gq-Rho regulation of locomotion, but that the canonical ERK activator LET-60/Ras may not be required. Through neuron-specific rescue experiments, we found that the ERK pathway functions in head acetylcholine neurons to control Gq-dependent locomotion. Additionally, expression of activated LIN-45/Raf in head acetylcholine neurons is sufficient to cause an exaggerated waveform phenotype and hypersensitivity to the acetylcholinesterase inhibitor aldicarb, similar to an activated Gq mutant. Taken together, our results suggest that the ERK MAPK pathway modulates the output of Gq-Rho signaling to control locomotion behavior in C. elegans. KEYWORDS Gq signaling; ERK MAPK pathway; Rho small GTPase; C. elegans; G protein HE heterotrimeric G protein Gq is a conserved regulator of neurons, the Rho ortholog RHO-1 regulates synaptic activity Tneurotransmission in metazoans.