Association of Rare Missense Variants in the Second Intracellular Loop of Nav1.7 Sodium Channels with Familial Autism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Expression Profiling of Ion Channel Genes Predicts Clinical Outcome in Breast Cancer
UCSF UC San Francisco Previously Published Works Title Expression profiling of ion channel genes predicts clinical outcome in breast cancer Permalink https://escholarship.org/uc/item/1zq9j4nw Journal Molecular Cancer, 12(1) ISSN 1476-4598 Authors Ko, Jae-Hong Ko, Eun A Gu, Wanjun et al. Publication Date 2013-09-22 DOI http://dx.doi.org/10.1186/1476-4598-12-106 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Ko et al. Molecular Cancer 2013, 12:106 http://www.molecular-cancer.com/content/12/1/106 RESEARCH Open Access Expression profiling of ion channel genes predicts clinical outcome in breast cancer Jae-Hong Ko1, Eun A Ko2, Wanjun Gu3, Inja Lim1, Hyoweon Bang1* and Tong Zhou4,5* Abstract Background: Ion channels play a critical role in a wide variety of biological processes, including the development of human cancer. However, the overall impact of ion channels on tumorigenicity in breast cancer remains controversial. Methods: We conduct microarray meta-analysis on 280 ion channel genes. We identify candidate ion channels that are implicated in breast cancer based on gene expression profiling. We test the relationship between the expression of ion channel genes and p53 mutation status, ER status, and histological tumor grade in the discovery cohort. A molecular signature consisting of ion channel genes (IC30) is identified by Spearman’s rank correlation test conducted between tumor grade and gene expression. A risk scoring system is developed based on IC30. We test the prognostic power of IC30 in the discovery and seven validation cohorts by both Cox proportional hazard regression and log-rank test. -

Autosomal Recessive Ataxias and with the Early-Onset Parkinson’S Disease That We Identified Here As Overlapping with the SFARI Autism Gene
Roadmap to Help Develop Personalized Targeted Treatments for Autism as a Disorder of the Nervous Systems Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) 1 | P a g e Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -

Characterizing Nav1.5 Expression, Organization, and Electrical Behavior in Cardiomyocyte Domains
Graduate School for Cellular and Biomedical Sciences University of Bern Characterizing Nav1.5 expression, organization, and electrical behavior in cardiomyocyte domains PhD Thesis submitted by Sarah Helena Vermij from the Netherlands for the degree of PhD in Biomedical Sciences Supervisor Prof. Dr. Hugues Abriel Institute of Biochemistry and Molecular Medicine Faculty of Medicine of the University of Bern Co-advisor Prof. Dr. Stephan Rohr Department of Physiology Faculty of Medicine of the University of Bern Urheberrechtlicher Hinweis Dieses Dokument steht unter einer Lizenz der Creative Commons Namensnennung-Keine kommerzielle Nutzung-Keine Bearbeitung 2.5 Schweiz. http://creativecommons.org/licenses/by-nc-nd/2.5/ch/ Sie dürfen: dieses Werk vervielfältigen, verbreiten und öffentlich zugänglich machen Zu den folgenden Bedingungen: Namensnennung. Sie müssen den Namen des Autors/Rechteinhabers in der von ihm festgelegten Weise nennen (wodurch aber nicht der Eindruck entstehen darf, Sie oder die Nutzung des Werkes durch Sie würden entlohnt). Keine kommerzielle Nutzung. Dieses Werk darf nicht für kommerzielle Zwecke verwendet werden. Keine Bearbeitung. Dieses Werk darf nicht bearbeitet oder in anderer Weise verändert werden. Im Falle einer Verbreitung müssen Sie anderen die Lizenzbedingungen, unter welche dieses Werk fällt, mitteilen. Jede der vorgenannten Bedingungen kann aufgehoben werden, sofern Sie die Einwilligung des Rechteinhabers dazu erhalten. Diese Lizenz lässt die Urheberpersönlichkeitsrechte nach Schweizer Recht unberührt. Eine ausführliche Fassung des Lizenzvertrags befindet sich unter http://creativecommons.org/licenses/by-nc-nd/2.5/ch/legalcode.de Accepted by the Faculty of Medicine, the Faculty of Science and the Vetsuisse Faculty of the University of Bern at the request of the Graduate School for Cellular and Biomedical Sciences Bern, Dean of the Faculty of Medicine Bern, Dean of the Faculty of Science Bern, Dean of the Vetsuisse Faculty Bern ABSTRACT Proper function of the heart depends on the function of voltage-gated ion channels. -

Identification of Key Genes and Pathways Involved in Response To
Deng et al. Biol Res (2018) 51:25 https://doi.org/10.1186/s40659-018-0174-7 Biological Research RESEARCH ARTICLE Open Access Identifcation of key genes and pathways involved in response to pain in goat and sheep by transcriptome sequencing Xiuling Deng1,2†, Dong Wang3†, Shenyuan Wang1, Haisheng Wang2 and Huanmin Zhou1* Abstract Purpose: This aim of this study was to investigate the key genes and pathways involved in the response to pain in goat and sheep by transcriptome sequencing. Methods: Chronic pain was induced with the injection of the complete Freund’s adjuvant (CFA) in sheep and goats. The animals were divided into four groups: CFA-treated sheep, control sheep, CFA-treated goat, and control goat groups (n 3 in each group). The dorsal root ganglions of these animals were isolated and used for the construction of a cDNA= library and transcriptome sequencing. Diferentially expressed genes (DEGs) were identifed in CFA-induced sheep and goats and gene ontology (GO) enrichment analysis was performed. Results: In total, 1748 and 2441 DEGs were identifed in CFA-treated goat and sheep, respectively. The DEGs identi- fed in CFA-treated goats, such as C-C motif chemokine ligand 27 (CCL27), glutamate receptor 2 (GRIA2), and sodium voltage-gated channel alpha subunit 3 (SCN3A), were mainly enriched in GO functions associated with N-methyl- D-aspartate (NMDA) receptor, infammatory response, and immune response. The DEGs identifed in CFA-treated sheep, such as gamma-aminobutyric acid (GABA)-related DEGs (gamma-aminobutyric acid type A receptor gamma 3 subunit [GABRG3], GABRB2, and GABRB1), SCN9A, and transient receptor potential cation channel subfamily V member 1 (TRPV1), were mainly enriched in GO functions related to neuroactive ligand-receptor interaction, NMDA receptor, and defense response. -

The Methamphetamine-Induced RNA Targetome of Hnrnp H in Hnrnph1 Mutants Showing Reduced Dopamine Release and Behavior
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.06.451358; this version posted July 7, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The methamphetamine-induced RNA targetome of hnRNP H in Hnrnph1 mutants showing reduced dopamine release and behavior Qiu T. Ruan1,2,3, Michael A. Rieger4, William B. Lynch2,5, Jiayi W. Cox6, Jacob A. Beierle1,2,3, Emily J. Yao2, Amarpreet Kandola2, Melanie M. Chen2, Julia C. Kelliher2, Richard K. Babbs2, Peter E. A. Ash7, Benjamin Wolozin7, Karen K. Szumlinski8, W. Evan Johnson9, Joseph D. Dougherty4, and Camron D. Bryant1,2,3* 1. Biomolecular Pharmacology Training Program, Department of Pharmacology and Experimental Therapeutics, Boston University School of Medicine 2. Laboratory of Addiction Genetics, Department of Pharmacology and Experimental Therapeutics and Psychiatry, Boston University School of Medicine 3. Transformative Training Program in Addiction Science, Boston University School of Medicine 4. Department of Genetics, Department of Psychiatry, Washington University School of Medicine 5. Graduate Program for Neuroscience, Boston University 6. Programs in Biomedical Sciences, Boston University School of Medicine 7. Department of Pharmacology and Experimental Therapeutics and Neurology, Boston University School of Medicine 8. Department of Psychological and Brain Sciences, University of California, Santa Barbara 9. Department of Medicine, Computational Biomedicine, Boston University School of Medicine *Corresponding Author Camron D. Bryant, Ph.D. Department of Pharmacology and Experimental Therapeutics and Department of Psychiatry 72 E. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

Ageing in Pgc-1Β Mice Modelling Mitochondrial Dysfunction Induces
Bioscience Reports (2019) 39 BSR20190127 https://doi.org/10.1042/BSR20190127 Research Article Ageing in Pgc-1β−/− mice modelling mitochondrial dysfunction induces differential expression of a range of genes regulating ventricular electrophysiology Downloaded from http://portlandpress.com/bioscirep/article-pdf/39/4/BSR20190127/842558/bsr-2019-0127-t.pdf by guest on 05 October 2020 Charlotte E. Edling1, Ibrahim T. Fazmin1,2, Karan R. Chadda1,2, Shiraz Ahmad2, Haseeb Valli2, Andrew A. Grace3, Christopher L.-H. Huang2,3 and Kamalan Jeevaratnam1,2,4 1Faculty of Health and Medical Sciences, University of Surrey, Guildford GU2 7AL, United Kingdom; 2Physiological Laboratory, University of Cambridge, Downing Street, Cambridge CB2 3EG, United Kingdom; 3Department of Biochemistry, Hopkins Building, University of Cambridge, Cambridge CB2 1QW, United Kingdom; 4School of Medicine, Perdana University-Royal College of Surgeons Ireland 43400, Serdang Selangor Darul Ehsan, Malaysia Correspondence: Kamalan Jeevaratnam ([email protected]) Mice deficient in mitochondrial promoter peroxisome proliferator activated receptor-γ co-activator-1β (Pgc-1β−/−) is a valuable model for metabolic diseases and has been found to present with several pathologies including ventricular arrhythmia. In the present study, our aim was to shed light on the molecular mechanisms behind the observed arrhythmic substrate by studying how the expression of selected genes critical for cardiac function differs in wild-type (WT) compared with Pgc-1β knockout mice and young compared with aged mice. We found that a clear majority of genes are down-regulated in the Pgc-1β−/− ventricular tissue compared with the WT. Although most individual genes are not signifi- cantly differentially expressed, a pattern is apparent when the genes are grouped according to their functional properties. -
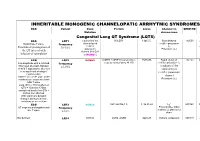
Inheritable Arrhytmia Syndromes
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT. -

Altered Physiological Functions and Ion Currents in Atrial Fibroblasts From
Physiological Reports ISSN 2051-817X ORIGINAL RESEARCH Altered physiological functions and ion currents in atrial fibroblasts from patients with chronic atrial fibrillation Claire Poulet1, Stephan Kunzel€ 1, Edgar Buttner€ 1, Diana Lindner2, Dirk Westermann2 & Ursula Ravens1 1 Department of Pharmacology and Toxicology, Medical Faculty Carl-Gustav-Carus, TU Dresden, Dresden, Germany 2 Department of General and Interventional Cardiology, University Heart Center Hamburg Eppendorf, Hamburg, Germany Keywords Abstract Atrial fibrillation, electrophysiology, fibroblasts. The contribution of human atrial fibroblasts to cardiac physiology and patho- physiology is poorly understood. Fibroblasts may contribute to arrhythmogen- Correspondence esis through fibrosis, or by directly altering electrical activity in Claire Poulet, Imperial College London, Imperial cardiomyocytes. The objective of our study was to uncover phenotypic differ- Centre for Translational and Experimental ences between cells from patients in sinus rhythm (SR) and chronic atrial fib- Medicine, Hammersmith Campus, Du Cane rillation (AF), with special emphasis on electrophysiological properties. We Road, London W12 0NN, UK isolated fibroblasts from human right atrial tissue for patch-clamp experi- Tel: +44 207 594 2738 Fax: +44 207 594 3653 ments, proliferation, migration, and differentiation assays, and gene expression E-mail: [email protected] profiling. In culture, proliferation and migration of AF fibroblasts were strongly impaired but differentiation into myofibroblasts was increased. This Present Addresses was associated with a higher number of AF fibroblasts expressing functional Claire Poulet, Imperial College London, Nav1.5 channels. Strikingly Na+ currents were considerably larger in AF cells. National Heart and Lung Institute, London, UK Blocking Na+ channels in culture with tetrodotoxin did not affect prolifera- tion, migration, or differentiation in neither SR nor AF cells. -

Experimental Eye Research 129 (2014) 93E106
Experimental Eye Research 129 (2014) 93e106 Contents lists available at ScienceDirect Experimental Eye Research journal homepage: www.elsevier.com/locate/yexer Transcriptomic analysis across nasal, temporal, and macular regions of human neural retina and RPE/choroid by RNA-Seq S. Scott Whitmore a, b, Alex H. Wagner a, c, Adam P. DeLuca a, b, Arlene V. Drack a, b, Edwin M. Stone a, b, Budd A. Tucker a, b, Shemin Zeng a, b, Terry A. Braun a, b, c, * Robert F. Mullins a, b, Todd E. Scheetz a, b, c, a Stephen A. Wynn Institute for Vision Research, The University of Iowa, Iowa City, IA, USA b Department of Ophthalmology and Visual Sciences, Carver College of Medicine, The University of Iowa, Iowa City, IA, USA c Department of Biomedical Engineering, College of Engineering, The University of Iowa, Iowa City, IA, USA article info abstract Article history: Proper spatial differentiation of retinal cell types is necessary for normal human vision. Many retinal Received 14 September 2014 diseases, such as Best disease and male germ cell associated kinase (MAK)-associated retinitis pigmen- Received in revised form tosa, preferentially affect distinct topographic regions of the retina. While much is known about the 31 October 2014 distribution of cell types in the retina, the distribution of molecular components across the posterior pole Accepted in revised form 4 November 2014 of the eye has not been well-studied. To investigate regional difference in molecular composition of Available online 5 November 2014 ocular tissues, we assessed differential gene expression across the temporal, macular, and nasal retina and retinal pigment epithelium (RPE)/choroid of human eyes using RNA-Seq. -

Title Human Genetic Diversity Possibly Determines Human's Compliance to COVID-19
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 16 April 2020 Title Human genetic diversity possibly determines human's compliance to COVID-19 1* 2 1 1 1 1 2 2 Yiqiang Zhao , Yongji Liu , Sa Li , Yuzhan Wang , Lina Bu , Qi Liu , Hongyi Lv , Fang Wan , Lida Wu2, Zeming Ning3, Yuchun Gu2,4* 1. Beijing Advanced Innovation Center for Food Nutrition and Human Health, China Agricultural University, Beijing, China 2. Allife Medicine Ltd., Beijing, China 3. The Wellcome Sanger Institute, Wellcome Genome Campus, Cambridge, UK 4. Regenerative Medicine Center, Aston University, Birmingham, UK Corresponding Author Prof. Yiqiang Zhao Beijing Advanced Innovation Center for Food Nutrition and Human Health China Agricultural University Beijing, China Email: [email protected] And Prof. Yuchun Gu The regenerative Medicine Center Aston University Birmingham, UK & Allife Medicine Co.Ltd Beijing, China Email: [email protected] © 2020 by the author(s). Distributed under a Creative Commons CC BY license. Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 16 April 2020 Abstract The rapid spread of the coronavirus disease 2019 (COVID-19) is a serious threat to public health systems globally and is subsequently, a cause of anxiety and panic within human society. Understanding the mechanisms and reducing the chances of having severe symptoms from COVID-19 will play an essential role in treating the disease, and become an urgent task to calm the panic. However, the COVID-19 test developed to identify virus carriers is unable to predict symptom development in individuals upon infection. Experiences from other plagues in human history and COVID-19 statistics suggest that genetic factors may determine the compliance with the virus, i.e., severe, mild, and asymptomatic.