Molecular Mapping of the Binding Sites of a Nav1.1 Activator
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Mineralocorticoid Receptor Leads to Increased Expression of EGFR
www.nature.com/scientificreports OPEN The mineralocorticoid receptor leads to increased expression of EGFR and T‑type calcium channels that support HL‑1 cell hypertrophy Katharina Stroedecke1,2, Sandra Meinel1,2, Fritz Markwardt1, Udo Kloeckner1, Nicole Straetz1, Katja Quarch1, Barbara Schreier1, Michael Kopf1, Michael Gekle1 & Claudia Grossmann1* The EGF receptor (EGFR) has been extensively studied in tumor biology and recently a role in cardiovascular pathophysiology was suggested. The mineralocorticoid receptor (MR) is an important efector of the renin–angiotensin–aldosterone‑system and elicits pathophysiological efects in the cardiovascular system; however, the underlying molecular mechanisms are unclear. Our aim was to investigate the importance of EGFR for MR‑mediated cardiovascular pathophysiology because MR is known to induce EGFR expression. We identifed a SNP within the EGFR promoter that modulates MR‑induced EGFR expression. In RNA‑sequencing and qPCR experiments in heart tissue of EGFR KO and WT mice, changes in EGFR abundance led to diferential expression of cardiac ion channels, especially of the T‑type calcium channel CACNA1H. Accordingly, CACNA1H expression was increased in WT mice after in vivo MR activation by aldosterone but not in respective EGFR KO mice. Aldosterone‑ and EGF‑responsiveness of CACNA1H expression was confrmed in HL‑1 cells by Western blot and by measuring peak current density of T‑type calcium channels. Aldosterone‑induced CACNA1H protein expression could be abrogated by the EGFR inhibitor AG1478. Furthermore, inhibition of T‑type calcium channels with mibefradil or ML218 reduced diameter, volume and BNP levels in HL‑1 cells. In conclusion the MR regulates EGFR and CACNA1H expression, which has an efect on HL‑1 cell diameter, and the extent of this regulation seems to depend on the SNP‑216 (G/T) genotype. -

Supplemental Table 1. Complete Gene Lists and GO Terms from Figure 3C
Supplemental Table 1. Complete gene lists and GO terms from Figure 3C. Path 1 Genes: RP11-34P13.15, RP4-758J18.10, VWA1, CHD5, AZIN2, FOXO6, RP11-403I13.8, ARHGAP30, RGS4, LRRN2, RASSF5, SERTAD4, GJC2, RHOU, REEP1, FOXI3, SH3RF3, COL4A4, ZDHHC23, FGFR3, PPP2R2C, CTD-2031P19.4, RNF182, GRM4, PRR15, DGKI, CHMP4C, CALB1, SPAG1, KLF4, ENG, RET, GDF10, ADAMTS14, SPOCK2, MBL1P, ADAM8, LRP4-AS1, CARNS1, DGAT2, CRYAB, AP000783.1, OPCML, PLEKHG6, GDF3, EMP1, RASSF9, FAM101A, STON2, GREM1, ACTC1, CORO2B, FURIN, WFIKKN1, BAIAP3, TMC5, HS3ST4, ZFHX3, NLRP1, RASD1, CACNG4, EMILIN2, L3MBTL4, KLHL14, HMSD, RP11-849I19.1, SALL3, GADD45B, KANK3, CTC- 526N19.1, ZNF888, MMP9, BMP7, PIK3IP1, MCHR1, SYTL5, CAMK2N1, PINK1, ID3, PTPRU, MANEAL, MCOLN3, LRRC8C, NTNG1, KCNC4, RP11, 430C7.5, C1orf95, ID2-AS1, ID2, GDF7, KCNG3, RGPD8, PSD4, CCDC74B, BMPR2, KAT2B, LINC00693, ZNF654, FILIP1L, SH3TC1, CPEB2, NPFFR2, TRPC3, RP11-752L20.3, FAM198B, TLL1, CDH9, PDZD2, CHSY3, GALNT10, FOXQ1, ATXN1, ID4, COL11A2, CNR1, GTF2IP4, FZD1, PAX5, RP11-35N6.1, UNC5B, NKX1-2, FAM196A, EBF3, PRRG4, LRP4, SYT7, PLBD1, GRASP, ALX1, HIP1R, LPAR6, SLITRK6, C16orf89, RP11-491F9.1, MMP2, B3GNT9, NXPH3, TNRC6C-AS1, LDLRAD4, NOL4, SMAD7, HCN2, PDE4A, KANK2, SAMD1, EXOC3L2, IL11, EMILIN3, KCNB1, DOK5, EEF1A2, A4GALT, ADGRG2, ELF4, ABCD1 Term Count % PValue Genes regulation of pathway-restricted GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, SMAD protein phosphorylation 9 6.34 1.31E-08 ENG pathway-restricted SMAD protein GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, phosphorylation -

Autosomal Recessive Ataxias and with the Early-Onset Parkinson’S Disease That We Identified Here As Overlapping with the SFARI Autism Gene
Roadmap to Help Develop Personalized Targeted Treatments for Autism as a Disorder of the Nervous Systems Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) 1 | P a g e Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -

Characterizing Nav1.5 Expression, Organization, and Electrical Behavior in Cardiomyocyte Domains
Graduate School for Cellular and Biomedical Sciences University of Bern Characterizing Nav1.5 expression, organization, and electrical behavior in cardiomyocyte domains PhD Thesis submitted by Sarah Helena Vermij from the Netherlands for the degree of PhD in Biomedical Sciences Supervisor Prof. Dr. Hugues Abriel Institute of Biochemistry and Molecular Medicine Faculty of Medicine of the University of Bern Co-advisor Prof. Dr. Stephan Rohr Department of Physiology Faculty of Medicine of the University of Bern Urheberrechtlicher Hinweis Dieses Dokument steht unter einer Lizenz der Creative Commons Namensnennung-Keine kommerzielle Nutzung-Keine Bearbeitung 2.5 Schweiz. http://creativecommons.org/licenses/by-nc-nd/2.5/ch/ Sie dürfen: dieses Werk vervielfältigen, verbreiten und öffentlich zugänglich machen Zu den folgenden Bedingungen: Namensnennung. Sie müssen den Namen des Autors/Rechteinhabers in der von ihm festgelegten Weise nennen (wodurch aber nicht der Eindruck entstehen darf, Sie oder die Nutzung des Werkes durch Sie würden entlohnt). Keine kommerzielle Nutzung. Dieses Werk darf nicht für kommerzielle Zwecke verwendet werden. Keine Bearbeitung. Dieses Werk darf nicht bearbeitet oder in anderer Weise verändert werden. Im Falle einer Verbreitung müssen Sie anderen die Lizenzbedingungen, unter welche dieses Werk fällt, mitteilen. Jede der vorgenannten Bedingungen kann aufgehoben werden, sofern Sie die Einwilligung des Rechteinhabers dazu erhalten. Diese Lizenz lässt die Urheberpersönlichkeitsrechte nach Schweizer Recht unberührt. Eine ausführliche Fassung des Lizenzvertrags befindet sich unter http://creativecommons.org/licenses/by-nc-nd/2.5/ch/legalcode.de Accepted by the Faculty of Medicine, the Faculty of Science and the Vetsuisse Faculty of the University of Bern at the request of the Graduate School for Cellular and Biomedical Sciences Bern, Dean of the Faculty of Medicine Bern, Dean of the Faculty of Science Bern, Dean of the Vetsuisse Faculty Bern ABSTRACT Proper function of the heart depends on the function of voltage-gated ion channels. -

An Advance About the Genetic Causes of Epilepsy
E3S Web of Conferences 271, 03068 (2021) https://doi.org/10.1051/e3sconf/202127103068 ICEPE 2021 An advance about the genetic causes of epilepsy Yu Sun1, a, *, †, Licheng Lu2, b, *, †, Lanxin Li3, c, *, †, Jingbo Wang4, d, *, † 1The School of Molecular and Cellular Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801-3633, US 2High School Affiliated to Shanghai Jiao Tong University, Shanghai, 200441, China 3Applied Biology program, University of British Columbia, Vancouver, V6r3b1, Canada 4School of Chemical Machinery and Safety, Dalian University of Technology, Dalian, 116023, China †These authors contributed equally. Abstract: Human hereditary epilepsy has been found related to ion channel mutations in voltage-gated channels (Na+, K+, Ca2+, Cl-), ligand gated channels (GABA receptors), and G-protein coupled receptors, such as Mass1. In addition, some transmembrane proteins or receptor genes, including PRRT2 and nAChR, and glucose transporter genes, such as GLUT1 and SLC2A1, are also about the onset of epilepsy. The discovery of these genetic defects has contributed greatly to our understanding of the pathology of epilepsy. This review focuses on introducing and summarizing epilepsy-associated genes and related findings in recent decades, pointing out related mutant genes that need to be further studied in the future. 1 Introduction Epilepsy is a neurological disorder characterized by 2 Malfunction of Ion channel epileptic seizures caused by abnormal brain activity. 1 in Functional variation in voltage or ligand-gated ion 100 (50 million people) people are affected by symptoms channel mutations is a major cause of idiopathic epilepsy, of this disorder worldwide, with men, young children, and especially in rare genetic forms. -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Ion Channels As Drug Targets
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central PERSPECTIVE Ion Channels as Drug Targets: The Next GPCRs Gregory J. Kaczorowski , Owen B. McManus, Birgit T. Priest , and Maria L. Garcia Department of Ion Channels, Merck Research Laboratories, Rahway, NJ 07065 Ion channels are well recognized as important thera- molecular target was unclear. Serendipity, insight, and peutic targets for treating a number of different patho- brute force effort drove these drug discovery efforts and physiologies. Historically, however, development of resulted in a number of notable successes including suc- drugs targeting this protein class has been diffi cult. Sev- cessful therapies and discovery of research tools that eral challenges associated with molecular-based drug have been invaluable in mapping out signaling path- discovery include validation of new channel targets and ways, purifying channel proteins, and characterizing identifi cation of acceptable medicinal chemistry leads. gating mechanisms, all of which has sustained the pres- Proof of concept approaches, focusing on combined ent era of ion channel drug discovery ( Garcia and molecular biological/pharmacological studies, have Kaczorowski, 2005 ). been successful. New, functional, high throughput With the advent of a more complete understanding screening (HTS) strategies developed to identify tracta- of cellular physiology and identifi cation of the molecu- ble lead structures, which typically are not abundant in lar components that constitute individual channel types small molecule libraries, have also yielded promising and control their function, researchers are now focus- results. Automated cell-based HTS assays can be confi g- ing on a molecular-based strategy to identify drugs tar- ured for many different types of ion channels using geting this protein class. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -
Inheritable Arrhytmia Syndromes
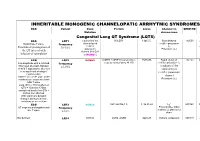
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT. -

Comparative Transcriptome Profiling of the Human and Mouse Dorsal Root Ganglia: an RNA-Seq-Based Resource for Pain and Sensory Neuroscience Research
bioRxiv preprint doi: https://doi.org/10.1101/165431; this version posted October 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq-based resource for pain and sensory neuroscience research Short Title: Human and mouse DRG comparative transcriptomics Pradipta Ray 1, 2 #, Andrew Torck 1 , Lilyana Quigley 1, Andi Wangzhou 1, Matthew Neiman 1, Chandranshu Rao 1, Tiffany Lam 1, Ji-Young Kim 1, Tae Hoon Kim 2, Michael Q. Zhang 2, Gregory Dussor 1 and Theodore J. Price 1, # 1 The University of Texas at Dallas, School of Behavioral and Brain Sciences 2 The University of Texas at Dallas, Department of Biological Sciences # Corresponding authors Theodore J Price Pradipta Ray School of Behavioral and Brain Sciences School of Behavioral and Brain Sciences The University of Texas at Dallas The University of Texas at Dallas BSB 14.102G BSB 10.608 800 W Campbell Rd 800 W Campbell Rd Richardson TX 75080 Richardson TX 75080 972-883-4311 972-883-7262 [email protected] [email protected] Number of pages: 27 Number of figures: 9 Number of tables: 8 Supplementary Figures: 4 Supplementary Files: 6 Word count: Abstract = 219; Introduction = 457; Discussion = 1094 Conflict of interest: The authors declare no conflicts of interest Patient anonymity and informed consent: Informed consent for human tissue sources were obtained by Anabios, Inc. (San Diego, CA). Human studies: This work was approved by The University of Texas at Dallas Institutional Review Board (MR 15-237). -

Pflugers Final
CORE Metadata, citation and similar papers at core.ac.uk Provided by Serveur académique lausannois A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Sylvain Pradervand2, Annie Mercier Zuber1, Gabriel Centeno1, Olivier Bonny1,3,4 and Dmitri Firsov1,4 1 - Department of Pharmacology and Toxicology, University of Lausanne, 1005 Lausanne, Switzerland 2 - DNA Array Facility, University of Lausanne, 1015 Lausanne, Switzerland 3 - Service of Nephrology, Lausanne University Hospital, 1005 Lausanne, Switzerland 4 – these two authors have equally contributed to the study to whom correspondence should be addressed: Dmitri FIRSOV Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925406 Fax: ++ 41-216925355 e-mail: [email protected] and Olivier BONNY Department of Pharmacology and Toxicology, University of Lausanne, 27 rue du Bugnon, 1005 Lausanne, Switzerland Phone: ++ 41-216925417 Fax: ++ 41-216925355 e-mail: [email protected] 1 Abstract The distal parts of the renal tubule play a critical role in maintaining homeostasis of extracellular fluids. In this review, we present an in-depth analysis of microarray-based gene expression profiles available for microdissected mouse distal nephron segments, i.e., the distal convoluted tubule (DCT) and the connecting tubule (CNT), and for the cortical portion of the collecting duct (CCD) (Zuber et al., 2009). Classification of expressed transcripts in 14 major functional gene categories demonstrated that all principal proteins involved in maintaining of salt and water balance are represented by highly abundant transcripts. However, a significant number of transcripts belonging, for instance, to categories of G protein-coupled receptors (GPCR) or serine-threonine kinases exhibit high expression levels but remain unassigned to a specific renal function.