A Biphasic Role for the Voltage-Gated Sodium Channel Scn5lab
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Protein Identities in Evs Isolated from U87-MG GBM Cells As Determined by NG LC-MS/MS
Protein identities in EVs isolated from U87-MG GBM cells as determined by NG LC-MS/MS. No. Accession Description Σ Coverage Σ# Proteins Σ# Unique Peptides Σ# Peptides Σ# PSMs # AAs MW [kDa] calc. pI 1 A8MS94 Putative golgin subfamily A member 2-like protein 5 OS=Homo sapiens PE=5 SV=2 - [GG2L5_HUMAN] 100 1 1 7 88 110 12,03704523 5,681152344 2 P60660 Myosin light polypeptide 6 OS=Homo sapiens GN=MYL6 PE=1 SV=2 - [MYL6_HUMAN] 100 3 5 17 173 151 16,91913397 4,652832031 3 Q6ZYL4 General transcription factor IIH subunit 5 OS=Homo sapiens GN=GTF2H5 PE=1 SV=1 - [TF2H5_HUMAN] 98,59 1 1 4 13 71 8,048185945 4,652832031 4 P60709 Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 - [ACTB_HUMAN] 97,6 5 5 35 917 375 41,70973209 5,478027344 5 P13489 Ribonuclease inhibitor OS=Homo sapiens GN=RNH1 PE=1 SV=2 - [RINI_HUMAN] 96,75 1 12 37 173 461 49,94108966 4,817871094 6 P09382 Galectin-1 OS=Homo sapiens GN=LGALS1 PE=1 SV=2 - [LEG1_HUMAN] 96,3 1 7 14 283 135 14,70620005 5,503417969 7 P60174 Triosephosphate isomerase OS=Homo sapiens GN=TPI1 PE=1 SV=3 - [TPIS_HUMAN] 95,1 3 16 25 375 286 30,77169764 5,922363281 8 P04406 Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 - [G3P_HUMAN] 94,63 2 13 31 509 335 36,03039959 8,455566406 9 Q15185 Prostaglandin E synthase 3 OS=Homo sapiens GN=PTGES3 PE=1 SV=1 - [TEBP_HUMAN] 93,13 1 5 12 74 160 18,68541938 4,538574219 10 P09417 Dihydropteridine reductase OS=Homo sapiens GN=QDPR PE=1 SV=2 - [DHPR_HUMAN] 93,03 1 1 17 69 244 25,77302971 7,371582031 11 P01911 HLA class II histocompatibility antigen, -

Transcriptomic Analysis of Native Versus Cultured Human and Mouse Dorsal Root Ganglia Focused on Pharmacological Targets Short
bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Transcriptomic analysis of native versus cultured human and mouse dorsal root ganglia focused on pharmacological targets Short title: Comparative transcriptomics of acutely dissected versus cultured DRGs Andi Wangzhou1, Lisa A. McIlvried2, Candler Paige1, Paulino Barragan-Iglesias1, Carolyn A. Guzman1, Gregory Dussor1, Pradipta R. Ray1,#, Robert W. Gereau IV2, # and Theodore J. Price1, # 1The University of Texas at Dallas, School of Behavioral and Brain Sciences and Center for Advanced Pain Studies, 800 W Campbell Rd. Richardson, TX, 75080, USA 2Washington University Pain Center and Department of Anesthesiology, Washington University School of Medicine # corresponding authors [email protected], [email protected] and [email protected] Funding: NIH grants T32DA007261 (LM); NS065926 and NS102161 (TJP); NS106953 and NS042595 (RWG). The authors declare no conflicts of interest Author Contributions Conceived of the Project: PRR, RWG IV and TJP Performed Experiments: AW, LAM, CP, PB-I Supervised Experiments: GD, RWG IV, TJP Analyzed Data: AW, LAM, CP, CAG, PRR Supervised Bioinformatics Analysis: PRR Drew Figures: AW, PRR Wrote and Edited Manuscript: AW, LAM, CP, GD, PRR, RWG IV, TJP All authors approved the final version of the manuscript. 1 bioRxiv preprint doi: https://doi.org/10.1101/766865; this version posted September 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Figure S1. Representative Report Generated by the Ion Torrent System Server for Each of the KCC71 Panel Analysis and Pcafusion Analysis
Figure S1. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. A Figure S1. Continued. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. B Figure S2. Comparative analysis of the variant frequency found by the KCC71 panel and calculated from publicly available cBioPortal datasets. For each of the 71 genes in the KCC71 panel, the frequency of variants was calculated as the variant number found in the examined cases. Datasets marked with different colors and sample numbers of prostate cancer are presented in the upper right. *Significantly high in the present study. Figure S3. Seven subnetworks extracted from each of seven public prostate cancer gene networks in TCNG (Table SVI). Blue dots represent genes that include initial seed genes (parent nodes), and parent‑child and child‑grandchild genes in the network. Graphical representation of node‑to‑node associations and subnetwork structures that differed among and were unique to each of the seven subnetworks. TCNG, The Cancer Network Galaxy. Figure S4. REVIGO tree map showing the predicted biological processes of prostate cancer in the Japanese. Each rectangle represents a biological function in terms of a Gene Ontology (GO) term, with the size adjusted to represent the P‑value of the GO term in the underlying GO term database. -

Genomic Diversity, Population Structure, and Signature of Selection in Five Chinese Native Sheep Breeds Adapted to Extreme Environments
G C A T T A C G G C A T genes Article Genomic Diversity, Population Structure, and Signature of Selection in Five Chinese Native Sheep Breeds Adapted to Extreme Environments Adam Abied 1,2 , Alnoor Bagadi 2, Farhad Bordbar 1, Yabin Pu 1, Serafino M.A. Augustino 3, Xianglan Xue 1 , Feng Xing 4, Gebremedhin Gebreselassie 1 , Jian-Lin Han 5,6 , Joram M. Mwacharo 7, Yuehui Ma 1 and Qianjun Zhao 1,* 1 Institute of Animal Science (IAS), Chinese Academy of Agricultural Sciences (CAAS), Beijing 100193, China; [email protected] (A.A.); [email protected] (F.B.); [email protected] (Y.P.); [email protected] (X.X.); [email protected] (G.G.); [email protected] (Y.M.) 2 Dry Land Research Center (DLRC) and Animal Production, Agricultural Research Corporation (ARC), Wad Madani 511, Sudan; [email protected] 3 College of Animal Science and Technology, China Agricultural University (CAU), Beijing 100193, China; serafi[email protected] 4 College of Animal Science, Talimu University (TU), Xinjiang, Alar 843300, China; [email protected] 5 CAAS-ILRI Joint Laboratory on Livestock and Forage Genetic Resources, Institute of Animal Science, Chinese Academy of Agricultural Sciences (CAAS), Beijing 100193, China; [email protected] 6 Livestock Genetics Program, International Livestock Research Institute (ILRI), Nairobi 00100, Kenya 7 International Center for Agricultural Research in the Dry Areas (ICARDA), Addis Ababa 1108-2010, Ethiopia; [email protected] * Correspondence: [email protected] Received: 27 February 2020; Accepted: 20 April 2020; Published: 30 April 2020 Abstract: Through long term natural and artificial selection, domestic sheep (Ovis aries) have become adapted to a diverse range of agro-ecological environments and display multiple phenotypic traits. -

Supplementary Figure S4
18DCIS 18IDC Supplementary FigureS4 22DCIS 22IDC C D B A E (0.77) (0.78) 16DCIS 14DCIS 28DCIS 16IDC 28IDC (0.43) (0.49) 0 ADAMTS12 (p.E1469K) 14IDC ERBB2, LASP1,CDK12( CCNE1 ( NUTM2B SDHC,FCGR2B,PBX1,TPR( CD1D, B4GALT3, BCL9, FLG,NUP21OL,TPM3,TDRD10,RIT1,LMNA,PRCC,NTRK1 0 ADAMTS16 (p.E67K) (0.67) (0.89) (0.54) 0 ARHGEF38 (p.P179Hfs*29) 0 ATG9B (p.P823S) (0.68) (1.0) ARID5B, CCDC6 CCNE1, TSHZ3,CEP89 CREB3L2,TRIM24 BRAF, EGFR (7p11); 0 ABRACL (p.R35H) 0 CATSPER1 (p.P152H) 0 ADAMTS18 (p.Y799C) 19q12 0 CCDC88C (p.X1371_splice) (0) 0 ADRA1A (p.P327L) (10q22.3) 0 CCNF (p.D637N) −4 −2 −4 −2 0 AKAP4 (p.G454A) 0 CDYL (p.Y353Lfs*5) −4 −2 Log2 Ratio Log2 Ratio −4 −2 Log2 Ratio Log2 Ratio 0 2 4 0 2 4 0 ARID2 (p.R1068H) 0 COL27A1 (p.G646E) 0 2 4 0 2 4 2 EDRF1 (p.E521K) 0 ARPP21 (p.P791L) ) 0 DDX11 (p.E78K) 2 GPR101, p.A174V 0 ARPP21 (p.P791T) 0 DMGDH (p.W606C) 5 ANP32B, p.G237S 16IDC (Ploidy:2.01) 16DCIS (Ploidy:2.02) 14IDC (Ploidy:2.01) 14DCIS (Ploidy:2.9) -3 -2 -1 -3 -2 -1 -3 -2 -1 -3 -2 -1 -3 -2 -1 -3 -2 -1 Log Ratio Log Ratio Log Ratio Log Ratio 12DCIS 0 ASPM (p.S222T) Log Ratio Log Ratio 0 FMN2 (p.G941A) 20 1 2 3 2 0 1 2 3 2 ERBB3 (p.D297Y) 2 0 1 2 3 20 1 2 3 0 ATRX (p.L1276I) 20 1 2 3 2 0 1 2 3 0 GALNT18 (p.F92L) 2 MAPK4, p.H147Y 0 GALNTL6 (p.E236K) 5 C11orf1, p.Y53C (10q21.2); 0 ATRX (p.R1401W) PIK3CA, p.H1047R 28IDC (Ploidy:2.0) 28DCIS (Ploidy:2.0) 22IDC (Ploidy:3.7) 22DCIS (Ploidy:4.1) 18IDC (Ploidy:3.9) 18DCIS (Ploidy:2.3) 17q12 0 HCFC1 (p.S2025C) 2 LCMT1 (p.S34A) 0 ATXN7L2 (p.X453_splice) SPEN, p.P677Lfs*13 CBFB 1 2 3 4 5 6 7 8 9 10 11 -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Autosomal Recessive Ataxias and with the Early-Onset Parkinson’S Disease That We Identified Here As Overlapping with the SFARI Autism Gene
Roadmap to Help Develop Personalized Targeted Treatments for Autism as a Disorder of the Nervous Systems Elizabeth B Torres 1,2,3* 1 Rutgers University Department of Psychology; [email protected] 2 Rutgers University Center for Cognitive Science (RUCCS) 3 Rutgers University Computer Science, Center for Biomedicine Imaging and Modelling (CBIM) * Correspondence: [email protected]; Tel.: (011) +858-445-8909 (E.B.T) 1 | P a g e Supplementary Material Sample Psychological criteria that sidelines sensory motor issues in autism: The ADOS-2 manual [1, 2], under the “Guidelines for Selecting a Module” section states (emphasis added): “Note that the ADOS-2 was developed for and standardized using populations of children and adults without significant sensory and motor impairments. Standardized use of any ADOS-2 module presumes that the individual can walk independently and is free of visual or hearing impairments that could potentially interfere with use of the materials or participation in specific tasks.” Sample Psychiatric criteria from the DSM-5 [3] that does not include sensory-motor issues: A. Persistent deficits in social communication and social interaction across multiple contexts, as manifested by the following, currently or by history (examples are illustrative, not exhaustive, see text): 1. Deficits in social-emotional reciprocity, ranging, for example, from abnormal social approach and failure of normal back-and-forth conversation; to reduced sharing of interests, emotions, or affect; to failure to initiate or respond to social interactions. 2. Deficits in nonverbal communicative behaviors used for social interaction, ranging, for example, from poorly integrated verbal and nonverbal communication; to abnormalities in eye contact and body language or deficits in understanding and use of gestures; to a total lack of facial expressions and nonverbal communication. -

Characterizing Nav1.5 Expression, Organization, and Electrical Behavior in Cardiomyocyte Domains
Graduate School for Cellular and Biomedical Sciences University of Bern Characterizing Nav1.5 expression, organization, and electrical behavior in cardiomyocyte domains PhD Thesis submitted by Sarah Helena Vermij from the Netherlands for the degree of PhD in Biomedical Sciences Supervisor Prof. Dr. Hugues Abriel Institute of Biochemistry and Molecular Medicine Faculty of Medicine of the University of Bern Co-advisor Prof. Dr. Stephan Rohr Department of Physiology Faculty of Medicine of the University of Bern Urheberrechtlicher Hinweis Dieses Dokument steht unter einer Lizenz der Creative Commons Namensnennung-Keine kommerzielle Nutzung-Keine Bearbeitung 2.5 Schweiz. http://creativecommons.org/licenses/by-nc-nd/2.5/ch/ Sie dürfen: dieses Werk vervielfältigen, verbreiten und öffentlich zugänglich machen Zu den folgenden Bedingungen: Namensnennung. Sie müssen den Namen des Autors/Rechteinhabers in der von ihm festgelegten Weise nennen (wodurch aber nicht der Eindruck entstehen darf, Sie oder die Nutzung des Werkes durch Sie würden entlohnt). Keine kommerzielle Nutzung. Dieses Werk darf nicht für kommerzielle Zwecke verwendet werden. Keine Bearbeitung. Dieses Werk darf nicht bearbeitet oder in anderer Weise verändert werden. Im Falle einer Verbreitung müssen Sie anderen die Lizenzbedingungen, unter welche dieses Werk fällt, mitteilen. Jede der vorgenannten Bedingungen kann aufgehoben werden, sofern Sie die Einwilligung des Rechteinhabers dazu erhalten. Diese Lizenz lässt die Urheberpersönlichkeitsrechte nach Schweizer Recht unberührt. Eine ausführliche Fassung des Lizenzvertrags befindet sich unter http://creativecommons.org/licenses/by-nc-nd/2.5/ch/legalcode.de Accepted by the Faculty of Medicine, the Faculty of Science and the Vetsuisse Faculty of the University of Bern at the request of the Graduate School for Cellular and Biomedical Sciences Bern, Dean of the Faculty of Medicine Bern, Dean of the Faculty of Science Bern, Dean of the Vetsuisse Faculty Bern ABSTRACT Proper function of the heart depends on the function of voltage-gated ion channels. -

Inhibition of ROMK Channels by Low Extracellular K and Oxidative Stress
Am J Physiol Renal Physiol 305: F208–F215, 2013. First published May 15, 2013; doi:10.1152/ajprenal.00185.2013. Inhibition of ROMK channels by low extracellular Kϩ and oxidative stress Gustavo Frindt,1 Hui Li,2 Henry Sackin,2 and Lawrence G. Palmer1 1Department of Physiology and Biophysics, Weill-Cornell Medical College, New York, New York; and 2Department of Physiology and Biophysics, The Chicago Medical School, Rosalind Franklin University, North Chicago, Illinois Submitted 2 April 2013; accepted in final form 8 May 2013 Frindt G, Li H, Sackin H, Palmer LG. Inhibition of ROMK may be essential for preventing Kϩ secretion and minimizing channels by low extracellular Kϩ and oxidative stress. Am J Physiol K losses. Renal Physiol 305: F208–F215, 2013. First published May 15, 2013; Measurements of ROMK activity in heterologous expression doi:10.1152/ajprenal.00185.2013.—We tested the hypothesis that low systems indicate that the channels are sensitive to changes in luminal Kϩ inhibits the activity of ROMK channels in the rat cortical ϩ ϩ ϩ the extracellular K concentration ([K ]o); decreases in [K ]o collecting duct. Whole-cell voltage-clamp measurements of the com- downregulate the channels (7, 28, 29, 31). One aspect of this Downloaded from ponent of outward Kϩ current inhibited by the bee toxin Tertiapin-Q ϩ response is a shift in the dependence of channel activity on (ISK) showed that reducing the bath concentration ([K ]o)to1mM intracellular pH, with low [Kϩ] moving the titration curve for resulted in a decline of current over 2 min compared with that o ϩ inhibition of the channels toward a higher, more physiological observed at 10 mM [K ]o. -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -
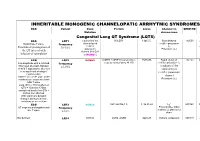
Inheritable Arrhytmia Syndromes
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT.