Studies on the Regulatory Roles of Cholesterol and Bile Acids
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae
Journal of Fungi Article Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae Mohammad Sayari 1,2,*, Magrieta A. van der Nest 1,3, Emma T. Steenkamp 1, Saleh Rahimlou 4 , Almuth Hammerbacher 1 and Brenda D. Wingfield 1 1 Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0002, South Africa; [email protected] (M.A.v.d.N.); [email protected] (E.T.S.); [email protected] (A.H.); brenda.wingfi[email protected] (B.D.W.) 2 Department of Plant Science, University of Manitoba, 222 Agriculture Building, Winnipeg, MB R3T 2N2, Canada 3 Biotechnology Platform, Agricultural Research Council (ARC), Onderstepoort Campus, Pretoria 0110, South Africa 4 Department of Mycology and Microbiology, University of Tartu, 14A Ravila, 50411 Tartu, Estonia; [email protected] * Correspondence: [email protected]; Fax: +1-204-474-7528 Abstract: Terpenes represent the biggest group of natural compounds on earth. This large class of organic hydrocarbons is distributed among all cellular organisms, including fungi. The different classes of terpenes produced by fungi are mono, sesqui, di- and triterpenes, although triterpene ergosterol is the main sterol identified in cell membranes of these organisms. The availability of genomic data from members in the Ceratocystidaceae enabled the detection and characterization of the genes encoding the enzymes in the mevalonate and ergosterol biosynthetic pathways. Using Citation: Sayari, M.; van der Nest, a bioinformatics approach, fungal orthologs of sterol biosynthesis genes in nine different species M.A.; Steenkamp, E.T.; Rahimlou, S.; of the Ceratocystidaceae were identified. -

Aquatic Toxicology 176 (2016) 116–127
Aquatic Toxicology 176 (2016) 116–127 Contents lists available at ScienceDirect Aquatic Toxicology j ournal homepage: www.elsevier.com/locate/aquatox Linking the response of endocrine regulated genes to adverse effects on sex differentiation improves comprehension of aromatase inhibition in a Fish Sexual Development Test a,∗ b b b Elke Muth-Köhne , Kathi Westphal-Settele , Jasmin Brückner , Sabine Konradi , c a a,1 d,1 Viktoria Schiller , Christoph Schäfers , Matthias Teigeler , Martina Fenske a Fraunhofer IME, Department of Ecotoxicology, Auf dem Aberg 1, 57392 Schmallenberg, Germany b German Environment Agency (UBA), Woerlitzer Platz 1, 06844 Dessau, Germany c Fraunhofer IME, Attract Group UNIFISH, Forckenbeckstraße 6, 52074 Aachen, Germany d Fraunhofer IME, Project Group Translational Medicine and Pharmacology TMP, Forckenbeckstraße 6, 52074 Aachen, Germany a r a t i c l e i n f o b s t r a c t Article history: The Fish Sexual Development Test (FSDT) is a non-reproductive test to assess adverse effects of endocrine Received 23 December 2015 disrupting chemicals. With the present study it was intended to evaluate whether gene expression end- Received in revised form 13 April 2016 points would serve as predictive markers of endocrine disruption in a FSDT. For proof-of-concept, a FSDT Accepted 19 April 2016 according to the OECD TG 234 was conducted with the non-steroidal aromatase inhibitor fadrozole (test Available online 20 April 2016 concentrations: 10 g/L, 32 g/L, 100 g/L) using zebrafish (Danio rerio). Gene expression analyses using quantitative RT-PCR were included at 48 h, 96 h, 28 days and 63 days post fertilization (hpf, dpf). -

Steroidal Triterpenes of Cholesterol Synthesis
Molecules 2013, 18, 4002-4017; doi:10.3390/molecules18044002 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Steroidal Triterpenes of Cholesterol Synthesis Jure Ačimovič and Damjana Rozman * Centre for Functional Genomics and Bio-Chips, Faculty of Medicine, Institute of Biochemistry, University of Ljubljana, Zaloška 4, Ljubljana SI-1000, Slovenia; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +386-1-543-7591; Fax: +386-1-543-7588. Received: 18 February 2013; in revised form: 19 March 2013 / Accepted: 27 March 2013 / Published: 4 April 2013 Abstract: Cholesterol synthesis is a ubiquitous and housekeeping metabolic pathway that leads to cholesterol, an essential structural component of mammalian cell membranes, required for proper membrane permeability and fluidity. The last part of the pathway involves steroidal triterpenes with cholestane ring structures. It starts by conversion of acyclic squalene into lanosterol, the first sterol intermediate of the pathway, followed by production of 20 structurally very similar steroidal triterpene molecules in over 11 complex enzyme reactions. Due to the structural similarities of sterol intermediates and the broad substrate specificity of the enzymes involved (especially sterol-Δ24-reductase; DHCR24) the exact sequence of the reactions between lanosterol and cholesterol remains undefined. This article reviews all hitherto known structures of post-squalene steroidal triterpenes of cholesterol synthesis, their biological roles and the enzymes responsible for their synthesis. Furthermore, it summarises kinetic parameters of enzymes (Vmax and Km) and sterol intermediate concentrations from various tissues. Due to the complexity of the post-squalene cholesterol synthesis pathway, future studies will require a comprehensive meta-analysis of the pathway to elucidate the exact reaction sequence in different tissues, physiological or disease conditions. -

Expression of Selected Genes Involved in Steroidogenesis in the Course of Enucleation-Induced Rat Adrenal Regeneration
INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 33: 613-623, 2014 Expression of selected genes involved in steroidogenesis in the course of enucleation-induced rat adrenal regeneration MARIANNA TYCZEWSKA, MARCIN RUCINSKI, AGNIESZKA ZIOLKOWSKA, MARCIN TREJTER, MARTA SZYSZKA and LUDWIK K. MALENDOWICZ Department of Histology and Embryology, Poznan University of Medical Sciences, Poznan, Poland Received October 28, 2013; Accepted December 6, 2013 DOI: 10.3892/ijmm.2013.1599 Abstract. The enucleation-induced (EI) rapid proliferation however, throughout the entire experimental period, there were of adrenocortical cells is followed by their differentiation, no statistically significant differences observed. After the initial the degree of which may be characterized by the expression decrease in steroidogenic factor 1 (Sf-1) mRNA levels observed of genes directly and indirectly involved in steroid hormone on the 1st day of the experiment, a marked upregulation in its biosynthesis. In this study, out of 30,000 transcripts of genes expression was observed from there on. Data from the current identified by means of Affymetrix Rat Gene 1.1 ST Array, we study strongly suggest the role of Fabp6, Lipe and Soat1 in aimed to select genes (either up- or downregulated) involved supplying substrates of regenerating adrenocortical cells for in steroidogenesis in the course of enucleation-induced adrenal steroid synthesis. Our results indicate that during the first days regeneration. On day 1, we found 32 genes with altered of adrenal regeneration, intense synthesis of cholesterol may expression levels, 15 were upregulated and 17 were down- occur, which is then followed by its conversion into cholesteryl regulated [i.e., 3β-hydroxysteroid dehydrogenase (Hsd3β), esters. -
Figures Intermediary Ontogeny.Pptx
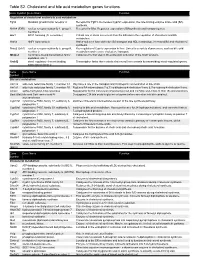
Table S2. Cholesterol and bile acid metabolism genes functions. Gene Symbol Gene Name Function Regulation of cholesterol and/or bile acid metabolism Fgfr4 fibroblast growth factor receptor 4 Receptor for Fgf15. Decreases Cyp7a1 expression, the rate-limiting enzyme in bile acid (BA) synthesis. Nr1h4 (FXR) nuclear receptor subfamily 1, group H, Receptor for BAs. Regulates expression of BAsynthesis and transport genes. member 4 Arv1 ARV1 homolog (S. cerevisiae) Critical role in sterol movement from the ER and in the regulation of cholesterol and BA metabolism. Hnf1a HNF1 homeobox A Hnf1a-null mice have defective BA transport and HDL metabolism, increased BA and cholesterol synthesis. Nr5a2 (Lrh1) nuclear receptor subfamily 5, group A, Key regulator of Cyp7a expression in liver. Linked to a variety of processes, such as bile acid member 2 metabolism and reverse cholesterol transport. Mbtps1 membrane-bound transcription factor Catalyzes the first step in the proteolytic activation of the Srebf proteins. peptidase, site 1 Srebf2 sterol regulatory element binding Transcription factor that controls cholesterol homeostasis by transcribing sterol-regulated genes. transcription factor 2 Gene Gene Name Function Symbol Bile acid metabolism Akr1c6 aldo-keto reductase family 1, member C1 May have a role in the transport and intrahepatic concentration of bile acids. Akr1d1 aldo-keto reductase family 1, member D1 Reduces BA intermediates 7-α,12-α-dihydroxy-4-cholesten-3-one & 7-α-hydroxy-4-cholesten-3-one. Amacr alpha-methylacyl-CoA racemase Responsible for the conversion of pristanoyl-CoA and C27-bile acyl-CoAs to their (S)-stereoisomers. Baat (Bat) bile acid CoA: amino acid N- Conjugates C24 bile acids to glycine or taurine before excretion into bile canaliculi. -

SUPPLEMENTARY MATERIAL Supplementary Table 1
Predicting Drug Promiscuity Using Spherical Harmonic Surface The Open Conference Proceedings Journal, 2011, Volume 2 i SUPPLEMENTARY MATERIAL Supplementary Table 1. MDDR Target Annotations Used in the Promiscuity Predictions TARGET_KEY EXTERNAL_ID 1001 prostaglandin-endoperoxide synthase 1002 cyclooxygenase 1 1003 cyclooxygenase 2 1046 ribonucleoside-diphosphate reductase 1112 thymidylate synthase 1209 phosphoribosylglycinamide formyltransferase 1609 purine-nucleoside phosphorylase 1611 uridine phosphorylase 1637 nad adp-ribosyltransferase 1657 transferring alkyl or aryl groups, other than methyl groups 1798 thymidine kinase 1814 protein kinase 1843 1-phosphatidylinositol 4-kinase 1883 protein-tyrosine kinase 1973 nucleotidyltransferase 2018 rna-directed dna polymerase 2121 phospholipase a2 2124 acetylcholinesterase 2282 phosphoric diester hydrolase 2283 phosphodiesterase i 2309 3',5'-cyclic-nucleotide phosphodiesterase 2310 3',5'-cyclic-gmp phosphodiesterase 2316 steryl-sulfatase 2414 alpha-glucosidase 2565 adenosylhomocysteinase 2576 peptidase 25 aldehyde reductase 2613 dipeptidyl-peptidase iv 2619 peptidyl-dipeptidase a 2624 serine-type d-ala-d-ala carboxypeptidase 2659 serine endopeptidase 2663 trypsin 2664 thrombin 2665 coagulation factor xa 2672 coagulation factor viia 2695 tryptase 2742 cathepsin b 2750 cathepsin l 2765 caspase-1 2784 hiv-1 retropepsin 2811 metalloendopeptidase 2822 stromelysin 1 286 xanthine oxidase 2978 beta-lactamase 3014 adenosine deaminase ii The Open Conference Proceedings Journal, 2011, Volume 2 Pérez-Nueno -

The Use of Mutants and Inhibitors to Study Sterol Biosynthesis in Plants
bioRxiv preprint doi: https://doi.org/10.1101/784272; this version posted September 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Title page 2 Title: The use of mutants and inhibitors to study sterol 3 biosynthesis in plants 4 5 Authors: Kjell De Vriese1,2, Jacob Pollier1,2,3, Alain Goossens1,2, Tom Beeckman1,2, Steffen 6 Vanneste1,2,4,* 7 Affiliations: 8 1: Department of Plant Biotechnology and Bioinformatics, Ghent University, Technologiepark 71, 9052 Ghent, 9 Belgium 10 2: VIB Center for Plant Systems Biology, VIB, Technologiepark 71, 9052 Ghent, Belgium 11 3: VIB Metabolomics Core, Technologiepark 71, 9052 Ghent, Belgium 12 4: Lab of Plant Growth Analysis, Ghent University Global Campus, Songdomunhwa-Ro, 119, Yeonsu-gu, Incheon 13 21985, Republic of Korea 14 15 e-mails: 16 K.D.V: [email protected] 17 J.P: [email protected] 18 A.G. [email protected] 19 T.B. [email protected] 20 S.V. [email protected] 21 22 *Corresponding author 23 Tel: +32 9 33 13844 24 Date of submission: sept 26th 2019 25 Number of Figures:3 in colour 26 Word count: 6126 27 28 1 bioRxiv preprint doi: https://doi.org/10.1101/784272; this version posted September 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Oxygen-Mediated Regulation of Cholesterol Synthesis
OXYGEN-MEDIATED REGULATION OF CHOLESTEROL SYNTHESIS THROUGH ACCELERATED DEGRADATION OF HMG COA REDUCTASE APPROVED BY SUPERVISORY COMMITTEE Russell DeBose-Boyd, Ph.D. Richard Bruick, Ph.D. Michael Brown, M.D. Zhijian Chen, Ph.D. George DeMartino, Ph.D. To my family For their love and support OXYGEN-MEDIATED REGULATION OF CHOLESTEROL SYNTHESIS THROUGH ACCELERATED DEGRADATION OF HMG COA REDUCTASE by ANDREW TUAN DUC NGUYEN DISSERTATION Presented to the Faculty of the Graduate School of Biomedical Sciences The University of Texas Southwestern Medical Center at Dallas In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY The University of Texas Southwestern Medical Center at Dallas Dallas, Texas May, 2009 Copyright by Andrew Tuan Duc Nguyen, 2009 All Rights Reserved OXYGEN-MEDIATED REGULATION OF CHOLESTEROL SYNTHESIS THROUGH ACCELERATED DEGRADATION OF HMG COA REDUCTASE Andrew Tuan Duc Nguyen, Ph.D. The University of Texas Southwestern Medical Center at Dallas, 2009 Supervising Professor: Russell A. DeBose-Boyd, Ph.D. Endoplasmic reticulum-associated degradation of the enzyme 3-hydroxy-3- methylglutaryl CoA reductase represents one mechanism by which cholesterol synthesis is controlled in mammalian cells. The key reaction in this degradation is binding of reductase to Insig proteins in the endoplasmic reticulum, which is stimulated by the methylated cholesterol precursors lanosterol and 24,25-dihydrolanosterol. Conversion of these sterols to cholesterol requires the removal of three methyl groups, which consumes v nine molecules of O2. Here, we report that oxygen deprivation (hypoxia) slows the rate of demethylation of lanosterol and its reduced metabolite 24,25-dihydrolanosterol, causing both sterols to accumulate in cells. -

In-Silico Screening of Potential Inhibitors of Hmgcoa Reductase and Lanosterol Synthase, Key Enzymes in Cholesterol Biosynthesis Pathway
In-silico screening of potential inhibitors of HMGCoA reductase and Lanosterol synthase, key enzymes in Cholesterol biosynthesis pathway A thesis submitted in partial fulfilment of the requirements for the degree of Bachelor of Technology In Biotechnology By N SAI VENKATA SARATH CHANDRA (110BT0626) Department of Biotechnology and Medical Engineering National Institute of Technology, Rourkela Rourkela, Odisha 769008, India May 2014 Page | CERTIFICATE This is to certify that the project thesis report entitled “In-silico screening of potential inhibitors of HMGCoA reductase and Lanosterol synthase, key enzymes in Cholesterol biosynthesis pathway” submitted by N SAI VENKATA SARATH CHANDRA (110BT0626) in the partial fulfilment of the requirement for the degree of the B.Tech in Biotechnology in Department of Biotechnology and Medical Engineering, National Institute of Technology, Rourkela is a record of an authentic bonafide research work carried out by him under my supervision and guidance. To the best of my knowledge, the matter embodied in the project thesis report has not been submitted to any other Institute/University for any Degree or Diploma. Dr. Nandini Sarkar (Supervisor) Date:- 12thMay2014 Assistant Professor Place:- Rourkela Department of Biotechnology and Medical Engineering National Institute of Technology, Rourkela Odisha-769008 Page | i ACKNOWLEDGEMENT I am glad to take this opportunity to express my heartfelt gratitude to my guide, Dr. Nandini Sarkar, without whose guidance my project would not have shaped well. I would thank her for her precious time, advices throughout my research effort and I also thank her for correcting my mistakes with her experience. I express my profound respect to Dr. Subhankar Paul, for helping me in the project and sparing his precious time. -

De Novo Androgen Synthesis As a Mechanism Contributing to the Progression of Prostate Cancer to Castration Resistance
DE NOVO ANDROGEN SYNTHESIS AS A MECHANISM CONTRIBUTING TO THE PROGRESSION OF PROSTATE CANCER TO CASTRATION RESISTANCE by JENNIFER ANN LOCKE B.Sc., The University of British Columbia, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE STUDIES (Experimental Medicine) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) June 2009 © Jennifer Ann Locke, 2009 Abstract Prostate cancer (CaP) is the leading cause of cancer in men affecting 24,700 Canadians each year and the third leading cause of cancer mortality with 4,300 deaths each year. CaP cells are derived from the prostate secretory epithelium and depend on androgen ligand activation of androgen receptor (AR) for survival, growth and proliferation. Androgen deprivation therapy (ADT) through pharmacological methods has been the leading form of CaP therapy since Huggin‟s discovery that castration induced the regression of CaP tumors in 1941. Unfortunately, the cancer often recurs within 2-4 years in what has classically been considered “androgen-independent” (AI) disease. Growing evidence implicates androgens and AR activation in this disease recurrence despite castration, suggesting that this terminology should be more appropriately called “castration-resistant” prostate cancer (CRPC). Firstly, AR is found amplified, overexpressed or mutated in a majority of recurrent cancers as compared to primary cancers and secondly, intratumoral testosterone levels remain the same pre- and post-ADT. Additionally, the measured intratumoral DHT levels are sufficient to activate AR in recurrent CaP cells despite low serum androgen levels suggesting that intratumoral androgens remain important mediators of AR-mediated CaP progression. Previously, we and others discovered that recurrent tumor cells have elevated levels of enzymes in the pathways necessary for androgen synthesis from cholesterol. -

Hypercholesterolemia Is Recognized As a Risk Factor for Atherosclerotic Disease, Such As Coronary Heart Disease. Moderate Exerci
VOL. 56 NO. 10, OCT. 2003 THE JOURNAL OF ANTIBIOTICS pp. 817-826 Lanopylins A1, B1, A2 and B2, Novel Lanosterol Synthase Inhibitors from Streptomyces sp. K99-5041 YUICHI SAKANOa, MASAAKI SHIBUYAa, ATSUKO MATSUMOTOb,YOKO TAKAHASHIb, HIROSHI TOMODAb, SATOSHI OMURAb and YUTAKA EBIZUKAa,* a Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan b Kitasato Institute for Life Sciences, Kitasato University, and The Kitasato Institute, 5-9-1 Shirokane, Minato-ku, Tokyo 108-8641, Japan (Received for publication June 25, 2003) From an actinomycete strain, Streptomyces sp. K99-5041, lanopylins A1, B1, A2 and B2 were isolated as new natural products that inhibited the reaction of recombinant human lanosterol synthase. The crude extract from the whole broth of this strain was fractionated by silica gel column chromatography to afford an active fraction that showed a single spot on TLC. Detailed analyses of this fraction with liquid chromatography-atmospheric pressure chemical ionization mass spectrometry revealed that it contained 20 homologous compounds with differing side chain lengths. The fraction was separated by preparative HPLC to afford four of these homologues, lanopylins A1, B1, A2 and B2. Detailed spectroscopic analyses of these isolated compounds led to the identification of their structures. Lanopylins A1 and B1 were (3E)-isohexadecylmethylidene-2-methyl-1-pyrroline and (3E)-hexadecylmethylidene-2-methyl- 1-pyrroline, respectively, and lanopylins A2 and B2 were homologues with the insertion of one cis-ethylenylidene in the side chain of lanopylins A1 and B1, respectively. These compounds inhibited recombinant human lanosterol synthase with IC50 values of 15, 18, 33, and 41μM, respectively. -

Combinatorial Biosynthesis of Sapogenins and Saponins in Saccharomyces Cerevisiae Using a C-16 Α Hydroxylase from Bupleurum
Combinatorial biosynthesis of sapogenins and saponins in Saccharomyces cerevisiae using a C-16α hydroxylase from Bupleurum falcatum Tessa Mosesa,b,c,d, Jacob Polliera,b, Lorena Almagroe, Dieter Buystf, Marc Van Montagua,b,1, María A. Pedreñoe, José C. Martinsf, Johan M. Theveleinc,d, and Alain Goossensa,b,1 aDepartment of Plant Systems Biology, VIB, 9052 Gent, Belgium; bDepartment of Plant Biotechnology and Bioinformatics, Ghent University, 9052 Gent, Belgium; cDepartment of Molecular Microbiology, VIB, 3001 Leuven-Heverlee, Belgium; dLaboratory of Molecular Cell Biology, Institute of Botany and Microbiology, Katholieke Universiteit Leuven, 3001 Leuven-Heverlee, Belgium; eDepartment of Plant Biology, Faculty of Biology, Universidad de Murcia, 30100 Murcia, Spain; and fDepartment of Organic Chemistry, Ghent University, 9000 Gent, Belgium Contributed by Marc Van Montagu, December 17, 2013 (sent for review April 29, 2013) The saikosaponins comprise oleanane- and ursane-type triterpene P450s that modify the β-amyrin backbone on C-11; C-12,13; saponins that are abundantly present in the roots of the genus C-16; C-22; C-23; C-28 or C-30 have been characterized from Bupleurum widely used in Asian traditional medicine. Here we Glycyrrhiza uralensis, Avena strigosa, Medicago truncatula, Glycine identified a gene, designated CYP716Y1, encoding a cytochrome max, Vitis vinifera,andCatharanthus roseus (11–18). Hydroxylases P450 monooxygenase from Bupleurum falcatum that catalyzes from Panax ginseng that oxidize the dammarenediol-II backbone the C-16α hydroxylation of oleanane- and ursane-type triterpenes. on C-6, C-12, or C-28 (19–21), and a C-20 hydroxylase from Lotus Exploiting this hitherto unavailable enzymatic activity, we launched japonicus (22) that modifies lupeol, have also been identified.