Host-Pathogen Genetic Interactions Underlie Tuberculosis Susceptibility in Genetically Diverse Mice [Preprint]
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

RHOBTB1 CRISPR/Cas9 KO Plasmid (H): Sc-418424
SANTA CRUZ BIOTECHNOLOGY, INC. RHOBTB1 CRISPR/Cas9 KO Plasmid (h): sc-418424 BACKGROUND APPLICATIONS The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and RHOBTB1 CRISPR/Cas9 KO Plasmid (h) is recommended for the disruption of CRISPR-associated protein (Cas9) system is an adaptive immune response gene expression in human cells. defense mechanism used by archea and bacteria for the degradation of foreign genetic material (4,6). This mechanism can be repurposed for other 20 nt non-coding RNA sequence: guides Cas9 functions, including genomic engineering for mammalian systems, such as to a specific target location in the genomic DNA gene knockout (KO) (1,2,3,5). CRISPR/Cas9 KO Plasmid products enable the U6 promoter: drives gRNA scaffold: helps Cas9 identification and cleavage of specific genes by utilizing guide RNA (gRNA) expression of gRNA bind to target DNA sequences derived from the Genome-scale CRISPR Knock-Out (GeCKO) v2 library developed in the Zhang Laboratory at the Broad Institute (3,5). Termination signal Green Fluorescent Protein: to visually REFERENCES verify transfection CRISPR/Cas9 Knockout Plasmid CBh (chicken β-Actin 1. Cong, L., et al. 2013. Multiplex genome engineering using CRISPR/Cas hybrid) promoter: drives expression of Cas9 systems. Science 339: 819-823. 2A peptide: allows production of both Cas9 and GFP from the 2. Mali, P., et al. 2013. RNA-guided human genome engineering via Cas9. same CBh promoter Science 339: 823-826. Nuclear localization signal 3. Ran, F.A., et al. 2013. Genome engineering using the CRISPR-Cas9 system. Nuclear localization signal SpCas9 ribonuclease Nat. Protoc. 8: 2281-2308. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

High Throughput Strategies Aimed at Closing the GAP in Our Knowledge of Rho Gtpase Signaling
cells Review High Throughput strategies Aimed at Closing the GAP in Our Knowledge of Rho GTPase Signaling Manel Dahmene 1, Laura Quirion 2 and Mélanie Laurin 1,3,* 1 Oncology Division, CHU de Québec–Université Laval Research Center, Québec, QC G1V 4G2, Canada; [email protected] 2 Montréal Clinical Research Institute (IRCM), Montréal, QC H2W 1R7, Canada; [email protected] 3 Université Laval Cancer Research Center, Québec, QC G1R 3S3, Canada * Correspondence: [email protected] Received: 21 May 2020; Accepted: 7 June 2020; Published: 9 June 2020 Abstract: Since their discovery, Rho GTPases have emerged as key regulators of cytoskeletal dynamics. In humans, there are 20 Rho GTPases and more than 150 regulators that belong to the RhoGEF, RhoGAP, and RhoGDI families. Throughout development, Rho GTPases choregraph a plethora of cellular processes essential for cellular migration, cell–cell junctions, and cell polarity assembly. Rho GTPases are also significant mediators of cancer cell invasion. Nevertheless, to date only a few molecules from these intricate signaling networks have been studied in depth, which has prevented appreciation for the full scope of Rho GTPases’ biological functions. Given the large complexity involved, system level studies are required to fully grasp the extent of their biological roles and regulation. Recently, several groups have tackled this challenge by using proteomic approaches to map the full repertoire of Rho GTPases and Rho regulators protein interactions. These studies have provided in-depth understanding of Rho regulators specificity and have contributed to expand Rho GTPases’ effector portfolio. Additionally, new roles for understudied family members were unraveled using high throughput screening strategies using cell culture models and mouse embryos. -

Open Questions: What About the 'Other' Rho Gtpases?
Ridley BMC Biology (2016) 14:64 DOI 10.1186/s12915-016-0289-7 COMMENT Open Access Open questions: what about the ‘other’ Rho GTPases? Anne J. Ridley Abstract know if they interact with and/or regulate the activity of other family members. Indeed, by studying only RhoA, Rho GTPases have many and diverse roles in cell Rac1 and Cdc42, we are likely to be missing the real physiology, and some family members are very well functions of many GEFs and GAPs because their targets studied, including RhoA, Rac1 and Cdc42. But many in cells are among the other Rho GTPases. are relatively neglected, and fundamental questions Interestingly, four family members—Rnd1, Rnd2, Rnd3 about their mechanisms and functions remain open. and RhoH—are ‘atypical’, in that they are known to be constitutively GTP-bound and do not hydrolyse GTP: much less is known about how these family members Rho GTPases are household names for anyone who are regulated. RhoU and RhoV have high intrinsic GDP/ works on eukaryotic cell migration and their functions GTP exchange rates, so are unlikely to need GEFs for in cell migration, cell division and cell polarity are de- activation but could still be turned off by GAPs [1, 4]. scribed in most textbooks on cell biology. Yet most of For two other members, RhoBTB1 and RhoBTB2, the what we know about Rho GTPases comes from studying Rho domains are quite divergent in sequence from other a small subset of the many different family members, family members and they are unlikely to be regulated by namely RhoA, Rac1 and Cdc42. -

Rho Guanine Nucleotide Exchange Factors: Regulators of Rho Gtpase Activity in Development and Disease
Oncogene (2014) 33, 4021–4035 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease DR Cook1, KL Rossman2,3 and CJ Der1,2,3 The aberrant activity of Ras homologous (Rho) family small GTPases (20 human members) has been implicated in cancer and other human diseases. However, in contrast to the direct mutational activation of Ras found in cancer and developmental disorders, Rho GTPases are activated most commonly in disease by indirect mechanisms. One prevalent mechanism involves aberrant Rho activation via the deregulated expression and/or activity of Rho family guanine nucleotide exchange factors (RhoGEFs). RhoGEFs promote formation of the active GTP-bound state of Rho GTPases. The largest family of RhoGEFs is comprised of the Dbl family RhoGEFs with 70 human members. The multitude of RhoGEFs that activate a single Rho GTPase reflects the very specific role of each RhoGEF in controlling distinct signaling mechanisms involved in Rho activation. In this review, we summarize the role of Dbl RhoGEFs in development and disease, with a focus on Ect2 (epithelial cell transforming squence 2), Tiam1 (T-cell lymphoma invasion and metastasis 1), Vav and P-Rex1/2 (PtdIns(3,4,5)P3 (phosphatidylinositol (3,4,5)-triphosphate)-dependent Rac exchanger). Oncogene (2014) 33, 4021–4035; doi:10.1038/onc.2013.362; published online 16 September 2013 Keywords: Rac1; RhoA; Cdc42; guanine nucleotide exchange factors; cancer; -

Post-Transcriptionally Impaired De Novo Mutations Contribute to The
bioRxiv preprint doi: https://doi.org/10.1101/175844; this version posted November 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Post-transcriptionally impaired de novo mutations 2 contribute to the genetic etiology of four neuropsychiatric 3 disorders 4 5 Fengbiao Mao1,2¶, Lu Wang3¶, Xiaolu Zhao2, Zhongshan Li4, Luoyuan Xiao5, 6 Rajesh C. Rao2, Jinchen Li4, Huajing Teng1*, Xin He6*, and Zhong Sheng Sun1,4* 7 8 1 Beijing Institutes of Life Science, Chinese Academy of Sciences, Beijing 100101, 9 China. 10 2 Department of Pathology, University of Michigan, Ann Arbor, MI 48109, USA. 11 3 Institute of Life Science, Southeast University, Nanjing 210096, China. 12 4 Institute of Genomic Medicine, Wenzhou Medical University, Wenzhou 325027, 13 China 14 5 Department of Computer Science and Technology, Tsinghua University, Beijing 15 100084, China. 16 6 Department of Human Genetics, University of Chicago, Chicago, IL, USA. 17 18 ¶These authors contributed equally to this work 19 * Corresponding authors 20 E-mail: 21 [email protected] (Z.S.S.) 22 [email protected] (X.H.) 23 [email protected] (H.T.) 24 25 1 bioRxiv preprint doi: https://doi.org/10.1101/175844; this version posted November 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Rho Gtpases of the Rhobtb Subfamily and Tumorigenesis1
Acta Pharmacol Sin 2008 Mar; 29 (3): 285–295 Invited review Rho GTPases of the RhoBTB subfamily and tumorigenesis1 Jessica BERTHOLD2, Kristína SCHENKOVÁ2, Francisco RIVERO2,3,4 2Centers for Biochemistry and Molecular Medicine, University of Cologne, Cologne, Germany; 3The Hull York Medical School, University of Hull, Hull HU6 7RX, UK Key words Abstract Rho guanosine triphosphatase; RhoBTB; RhoBTB proteins constitute a subfamily of atypical members within the Rho fa- DBC2; cullin; neoplasm mily of small guanosine triphosphatases (GTPases). Their most salient feature 1This work was supported by grants from the is their domain architecture: a GTPase domain (in most cases, non-functional) Center for Molecular Medicine Cologne, the is followed by a proline-rich region, a tandem of 2 broad-complex, tramtrack, Deutsche Forschungsgemeinschaft, and the bric à brac (BTB) domains, and a conserved C-terminal region. In humans, Köln Fortune Program of the Medical Faculty, University of Cologne. the RhoBTB subfamily consists of 3 isoforms: RhoBTB1, RhoBTB2, and RhoBTB3. Orthologs are present in several other eukaryotes, such as Drosophi- 4 Correspondence to Dr Francisco RIVERO. la and Dictyostelium, but have been lost in plants and fungi. Interest in RhoBTB Phn 49-221-478-6987. Fax 49-221-478-6979. arose when RHOBTB2 was identified as the gene homozygously deleted in E-mail [email protected] breast cancer samples and was proposed as a candidate tumor suppressor gene, a property that has been extended to RHOBTB1. The functions of RhoBTB pro- Received 2007-11-17 Accepted 2007-12-16 teins have not been defined yet, but may be related to the roles of BTB domains in the recruitment of cullin3, a component of a family of ubiquitin ligases. -
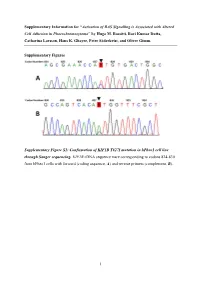
Activation of RAS Signalling Is Associated with Altered Cell Adhesion in Phaeochromocytoma” by Hugo M
Supplementary Information for “Activation of RAS Signalling is Associated with Altered Cell Adhesion in Phaeochromocytoma” by Hugo M. Rossitti, Ravi Kumar Dutta, Catharina Larsson, Hans K. Ghayee, Peter Söderkvist, and Oliver Gimm. Supplementary Figures Supplementary Figure S1: Confirmation of KIF1B T827I mutation in hPheo1 cell line through Sanger sequencing. KIF1B cDNA sequence trace corresponding to codons 824-830 from hPheo1 cells with forward (coding sequence, A) and reverse primers (complement, B). 1 Supplementary Figure S2: Confirmation of NRAS Q61K mutation in hPheo1 cell line through Sanger sequencing. NRAS cDNA sequence trace corresponding to codons 56-67 from hPheo1 cells with forward (coding sequence, A) and reverse primers (complement, B). 2 Supplementary Figure S3: CCND1 gene expression and hPheo1 proliferation. A: Expression of CCND1 mRNA assessed by RT-qPCR and presented as fold change (2-ΔΔCT, mean ± standard error of the mean). B: Cell counts at 1, 2, and 3 days after plating (corresponding to 72, 96 and 120 hours posttransfection, respectively) of control- or siNRAS#1-transfected hPheo1 cells expressed as fold change of the number of cells plated at day 0 (48 hours posttransfection; mean ± standard deviation). All results are from three independent siRNA experiments. 3 Supplementary Tables Supplementary Table S1: List of transcript cluster IDs significantly upregulated in hPheo1 by siNRAS treatment (comparison: siNRAS versus control-transfected hPheo1; ANOVA p < 0.05, FDR < 0.25, fold change < -1.5 or > 1.5). Transcript -

Atypical Rho Gtpases of the Rhobtb Subfamily: Roles in Vesicle Trafficking and Tumorigenesis
Review Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis Wei Ji 1 and Francisco Rivero 2,* 1 School of Life Science and Technology, Changchun University of Science and Technology, Changchun 130022, China; [email protected] 2 Centre for Cardiovascular and Metabolic Research, Hull York Medical School, University of Hull, Cottingham Road, Hull, East Riding of Yorkshire HU6 7RX, UK * Correspondence: [email protected]; Tel.: +44-1482466433 Academic Editor: Bor Luen Tang Received: 17 May 2016; Accepted: 8 June 2016; Published: date Abstract: RhoBTB proteins constitute a subfamily of atypical Rho GTPases represented in mammals by RhoBTB1, RhoBTB2, and RhoBTB3. Their characteristic feature is a carboxyl terminal extension that harbors two BTB domains capable of assembling cullin 3-dependent ubiquitin ligase complexes. The expression of all three RHOBTB genes has been found reduced or abolished in a variety of tumors. They are considered tumor suppressor genes and recent studies have strengthened their implication in tumorigenesis through regulation of the cell cycle and apoptosis. RhoBTB3 is also involved in retrograde transport from endosomes to the Golgi apparatus. One aspect that makes RhoBTB proteins atypical among the Rho GTPases is their proposed mechanism of activation. No specific guanine nucleotide exchange factors or GTPase activating proteins are known. Instead, RhoBTB might be activated through interaction with other proteins that relieve their auto-inhibited conformation and inactivated through auto-ubiquitination and destruction in the proteasome. In this review we discuss our current knowledge on the molecular mechanisms of action of RhoBTB proteins and the implications for tumorigenesis and other pathologic conditions. -

Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared Amongst Δγ T, Innate Lymphoid, and Th Cells
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 δγ is online at: average * The Journal of Immunology , 10 of which you can access for free at: 2016; 197:1460-1470; Prepublished online 6 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600643 http://www.jimmunol.org/content/197/4/1460 Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst T, Innate Lymphoid, and Th Cells You Jeong Lee, Gabriel J. Starrett, Seungeun Thera Lee, Rendong Yang, Christine M. Henzler, Stephen C. Jameson and Kristin A. Hogquist J Immunol cites 41 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2016/07/06/jimmunol.160064 3.DCSupplemental This article http://www.jimmunol.org/content/197/4/1460.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 26, 2021. The Journal of Immunology Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst gd T, Innate Lymphoid, and Th Cells You Jeong Lee,* Gabriel J. -

Murine Perinatal Beta Cell Proliferation and the Differentiation of Human Stem Cell Derived Insulin Expressing Cells Require NEUROD1
Page 1 of 105 Diabetes Murine perinatal beta cell proliferation and the differentiation of human stem cell derived insulin expressing cells require NEUROD1 Anthony I. Romer,1,2 Ruth A. Singer1,3, Lina Sui2, Dieter Egli,2* and Lori Sussel1,4* 1Department of Genetics and Development, Columbia University, New York, NY 10032, USA 2Department of Pediatrics, Columbia University, New York, NY 10032, USA 3Integrated Program in Cellular, Molecular and Biomedical Studies, Columbia University, New York, NY 10032, USA 4Department of Pediatrics, University of Colorado Denver School of Medicine, Denver, CO 80045, USA *Co-Corresponding Authors Dieter Egli 1150 St. Nicholas Avenue New York, NY 10032 [email protected] Lori Sussel 1775 Aurora Ct. Aurora, CO 80045 [email protected] Word Count: Abstract= 149; Body= 4773 Total Paper Figures= 7, Total Supplemental Tables= 4, Total Supplemental Figures= 5 Diabetes Publish Ahead of Print, published online September 13, 2019 Diabetes Page 2 of 105 Abstract Inactivation of the β cell transcription factor NEUROD1 causes diabetes in mice and humans. In this study, we uncovered novel functions of Neurod1 during murine islet cell development and during the differentiation of human embryonic stem cells (HESCs) into insulin-producing cells. In mice, we determined that Neurod1 is required for perinatal proliferation of alpha and beta cells. Surprisingly, apoptosis only makes a minor contribution to beta cell loss when Neurod1 is deleted. Inactivation of NEUROD1 in HESCs severely impaired their differentiation from pancreatic progenitors into insulin expressing (HESC-beta) cells; however survival or proliferation was not affected at the time points analyzed. NEUROD1 was also required in HESC-beta cells for the full activation of an essential beta cell transcription factor network. -

Transcriptome Profiling Reveals the Complexity of Pirfenidone Effects in IPF
ERJ Express. Published on August 30, 2018 as doi: 10.1183/13993003.00564-2018 Early View Original article Transcriptome profiling reveals the complexity of pirfenidone effects in IPF Grazyna Kwapiszewska, Anna Gungl, Jochen Wilhelm, Leigh M. Marsh, Helene Thekkekara Puthenparampil, Katharina Sinn, Miroslava Didiasova, Walter Klepetko, Djuro Kosanovic, Ralph T. Schermuly, Lukasz Wujak, Benjamin Weiss, Liliana Schaefer, Marc Schneider, Michael Kreuter, Andrea Olschewski, Werner Seeger, Horst Olschewski, Malgorzata Wygrecka Please cite this article as: Kwapiszewska G, Gungl A, Wilhelm J, et al. Transcriptome profiling reveals the complexity of pirfenidone effects in IPF. Eur Respir J 2018; in press (https://doi.org/10.1183/13993003.00564-2018). This manuscript has recently been accepted for publication in the European Respiratory Journal. It is published here in its accepted form prior to copyediting and typesetting by our production team. After these production processes are complete and the authors have approved the resulting proofs, the article will move to the latest issue of the ERJ online. Copyright ©ERS 2018 Copyright 2018 by the European Respiratory Society. Transcriptome profiling reveals the complexity of pirfenidone effects in IPF Grazyna Kwapiszewska1,2, Anna Gungl2, Jochen Wilhelm3†, Leigh M. Marsh1, Helene Thekkekara Puthenparampil1, Katharina Sinn4, Miroslava Didiasova5, Walter Klepetko4, Djuro Kosanovic3, Ralph T. Schermuly3†, Lukasz Wujak5, Benjamin Weiss6, Liliana Schaefer7, Marc Schneider8†, Michael Kreuter8†, Andrea Olschewski1,