A Chemoenzymatic Strategy for Protein-Nanocellulose Conjugates
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Structural Studies of Lumazine Synthases – Thermostability, Catalytic Mechanism and Molecular Assembly
Center for Structural Biochemistry Department of Biosciences at Novum Karolinska Institutet, S-141 57 Huddinge, Sweden Structural Studies of Lumazine Synthases – Thermostability, Catalytic Mechanism and Molecular Assembly Xiaofeng Zhang Stockholm 2005 Cover Illustration: Electron density of the active site of lumazine synthase from the hyperthermophilic bacterium Aquifex aeolicus. All previously published papers were reproduced with permission from the publisher. Published and printed by Karolinska University Press Box 200, SE-171 77 Stockholm, Sweden © Xiaofeng Zhang, 2005 ISBN 91-7140-605-0 ABSTRACT Riboflavin, also known as vitamin B2, is biosynthesized in plants, bacteria, archaea and fungi. The primary biological function of riboflavin is related to its existence as a component of the two coenzymes, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), which play an important role for electron transfer in energy metabolism. This project is mainly focused on structural studies of lumazine synthase (LS) from the hyperthermophilic bacterium Aquifex aeolicus (LSAQ). The enzyme is involved in the penultimate step of biosynthesis of riboflavin. The aim of this study is to gain insights into the structural basis of thermostability, catalytic mechanism as well as the molecular assembly of the enzyme. Methods used for these studies include X-ray crystallography, electron microscopy (EM), small angle X-ray scattering (SAXS) and differential scanning calorimetry (DSC). Lumazine synthase from the hyperthermophile A. aeolicus displays dramatic stability against high temperature. The calorimetric melting profile indicates an apparent melting temperature (Tm) of 120qC. The factors that determine the thermostability of A. aeolicus LS were revealed by structural comparisons (Paper I, 2001). In the second last step of riboflavin biosynthesis, lumazine synthase catalyzes the formation of 6-7-dimethyl-8-ribityllumazine, which is subsequently converted to riboflavin. -

CV for Mark Cushman
Mark Cushman CURRICULUM VITAE - MARK CUSHMAN Professional Title: Distinguished Professor of Medicinal Chemistry Date of Birth: August 20, 1945 Place of Birth: Fresno, California Citizenship: United States Education: Undergraduate: Fresno State College Fresno, California 1963-1965 Professional: Doctor of Pharmacy University of California San Francisco, California 1965-1969 Graduate: Ph.D., Pharmaceutical Chemistry University of California San Francisco, California 1969-1973 Postdoctoral: Organic Chemistry Massachusetts Institute of Technology Cambridge, Massachusetts 1973-1975 Academic Appointments: Assistant Professor of Medicinal Chemistry Purdue University, 1975-1980 Associate Professor of Medicinal Chemistry Purdue University, 1980-1985 Professor of Medicinal Chemistry Purdue University, 1985-2010 Distinguished Professor of Medicinal Chemistry Purdue University, 2010-present Adjunct Professor of Pharmacology and Toxicology Indiana University School of Medicine, Lafayette, 2014-present Sabbatical: Senior Fulbright Scholar Lehrstuhl für Organische Chemie und Biochemie der Technische Universität München Garching, West Germany, 1983-1984 Present Position: Distinguished Professor of Medicinal Chemistry Purdue University, 2012-present 1 Mark Cushman Awards and Honors: Bank of America Achievement Award in Music, 1963 University of California Regents Scholarship, 1965-1969 American Foundation for Pharmaceutical Education Fellowship, 1969-1973 National Defense Education Act Title IV Fellowship, 1969-1973 National Institutes of Health Postdoctoral -

Vitamin Biosynthesis As an Antifungal Target
Journal of Fungi Review Vitamin Biosynthesis as an Antifungal Target Zohar Meir and Nir Osherov * Department of Clinical Microbiology and Immunology, Sackler School of Medicine, Tel-Aviv University, Ramat-Aviv, Tel-Aviv 69978, Israel; [email protected] * Correspondence: [email protected]; Tel.: +972-3-640-9599; Fax: +972-3-640-9160 Received: 29 May 2018; Accepted: 13 June 2018; Published: 17 June 2018 Abstract: The large increase in the population of immunosuppressed patients, coupled with the limited efficacy of existing antifungals and rising resistance toward them, have dramatically highlighted the need to develop novel drugs for the treatment of invasive fungal infections. An attractive possibility is the identification of possible drug targets within essential fungal metabolic pathways not shared with humans. Here, we review the vitamin biosynthetic pathways (vitamins A–E, K) as candidates for the development of antifungals. We present a set of ranking criteria that identify the vitamin B2 (riboflavin), B5 (pantothenic acid), and B9 (folate) biosynthesis pathways as being particularly rich in new antifungal targets. We propose that recent scientific advances in the fields of drug design and fungal genomics have developed sufficiently to merit a renewed look at these pathways as promising sources for the development of novel classes of antifungals. Keywords: antifungals; fungal vitamin metabolism; drug target; essential genes 1. Introduction The number of life-threatening fungal infections has risen dramatically over the last twenty years. Recent estimates have identified a global burden of almost two million patients with systemic and invasive fungal infections, including ~700,000 cases of invasive candidiasis, ~500,000 cases of Pneumocystis jirovecii pneumonia, ~250,000 cases of invasive aspergillosis, ~220,000 cases of cryptococcal meningitis, and ~100,000 cases of disseminated histoplasmosis [1,2]. -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions. -
![Imidazo[4,5-B] Pyridines As Lumazine Synthase Inhibitors for Their Effective Antimicrobial Activity](https://docslib.b-cdn.net/cover/9111/imidazo-4-5-b-pyridines-as-lumazine-synthase-inhibitors-for-their-effective-antimicrobial-activity-2269111.webp)
Imidazo[4,5-B] Pyridines As Lumazine Synthase Inhibitors for Their Effective Antimicrobial Activity
Original Article In-silico docking based design and synthesis of [1H,3H] imidazo[4,5-b] pyridines as lumazine synthase inhibitors for their effective antimicrobial activity Sunil L. Harer, Manish S. Bhatia Department of ABSTRACT Pharmaceutical Chemistry, Purpose: The imidazopyridine moiety is important pharmacophore that has proven to be useful for a number Bharati Vidyapeeth College of Pharmacy, of biologically relevant targets, also reported to display antibacterial, antifungal, antiviral properties. Riboflavin Kolhapur, Maharashtra, biosynthesis involving catalytic step of Lumazine synthase is absent in animals and human, but present in India microorganism, one of marked advantage of this study. Still, this path is not exploited as antiinfective target. Here, we proposed different interactions between [1H,3H] imidazo[4,5‑b] pyridine test ligands and target protein Address for correspondence: Lumazine synthase (protein Data Bank 2C92), one‑step synthesis of title compounds and further evaluation of Prof. Sunil L. Harer, them for in vitro antimicrobial activity. Materials and Methods: Active pocket of the target protein involved in E-mail: sunil.harer5@gmail. com the interaction with the test ligands molecules was found using Biopredicta tools in VLifeMDS 4.3 Suite. In‑silico docking suggests H‑bonding, hydrophobic interaction, charge interaction, aromatic interaction, and Vanderwaal forces responsible for stabilizing enzyme‑inhibitor complex. Disc diffusion assay method was used for in vitro antimicrobial screening. Results and Discussion: Investigation of possible interaction between test ligands and target lumazine synthase of Mycobacterium tuberculosis suggested 1i and 2f as best fit candidates showing hydrogen bonding, hydrophobic, aromatic and Vanderwaal’s forces. Among all derivatives 1g, 1j, 1k, 1l, 2a, 2c, 2d, 2e, 2h, and 2j exhibited potent activities against bacteria and fungi compared to the standard Ciprofloxacin and Fluconazole, respectively. -

Structure-Guided Computational Approaches to Unravel Druggable Proteomic Landscape of Mycobacterium Leprae
fmolb-08-663301 May 3, 2021 Time: 16:56 # 1 REVIEW published: 07 May 2021 doi: 10.3389/fmolb.2021.663301 Structure-Guided Computational Approaches to Unravel Druggable Proteomic Landscape of Mycobacterium leprae Sundeep Chaitanya Vedithi1*†, Sony Malhotra2†, Marta Acebrón-García-de-Eulate1, Modestas Matusevicius1, Pedro Henrique Monteiro Torres3 and Tom L. Blundell1* 1 Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom, 2 Rutherford Appleton Laboratory, Science and Technology Facilities Council, Oxon, United Kingdom, 3 Laboratório de Modelagem e Dinâmica Molecular, Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil Leprosy, caused by Mycobacterium leprae (M. leprae), is treated with a multidrug Edited by: Alexandre G. De Brevern, regimen comprising Dapsone, Rifampicin, and Clofazimine. These drugs exhibit INSERM U1134 Biologie Intégrée du bacteriostatic, bactericidal and anti-inflammatory properties, respectively, and control Globule Rouge, France the dissemination of infection in the host. However, the current treatment is not cost- Reviewed by: effective, does not favor patient compliance due to its long duration (12 months) Stéphane Téletchéa, Université de Nantes, France and does not protect against the incumbent nerve damage, which is a severe Jeremy Esque, leprosy complication. The chronic infectious peripheral neuropathy associated with Institut Biotechnologique de Toulouse (INSA), France the disease is primarily due to the bacterial components infiltrating the Schwann *Correspondence: cells that protect neuronal axons, thereby inducing a demyelinating phenotype. There Sundeep Chaitanya Vedithi is a need to discover novel/repurposed drugs that can act as short duration and [email protected] effective alternatives to the existing treatment regimens, preventing nerve damage Tom L. -
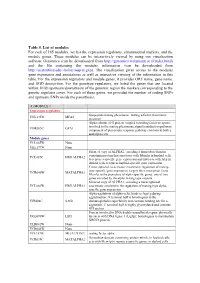
Table 5. List of Modules for Each of 165 Modules, We List the Expression Regulators, Chromosomal Markers, and the Module Genes
Table 5. List of modules For each of 165 modules, we list the expression regulators, chromosomal markers, and the module genes. These modules can be interactively viewed by using our visualization software Genomica (can be downloaded from http://genomica.weizmann.ac.il/index.html) and the file containing the modules information (can be downloaded from http://ai.stanford.edu/~koller/seqvar.gxp). The visualization gives access to the modules gene expression and annotations as well as interactive viewing of the information in this table. For the expression regulators and module genes, it provides ORF name, gene name, and SGD desciprtion. For the genotype regulators, we listed the genes that are located within 10 kb upstream/downstream of the genomic region the markers corresponding to the genetic regulator cover. For each of these genes, we provided the number of coding SNPs and upstream SNPs inside the parenthesis. (1) MODULE 1 Expression regulators lipopeptide mating pheromone; mating a-factor pheromone YNL145W MFA2 precursor Alpha subunit of G protein coupled to mating factor receptors, involved in the mating pheromone signal transduction pathway; YHR005C GPA1 component of pheromone response pathway common to both a and alpha cells Module genes YCL065W None YKL177W None Silenced copy of ALPHA2, encoding a homeobox-domain containing protein that associates with Mcm1p in haploid cells YCL067C HMLALPHA2 to repress a-specific gene expression and interacts with A1p in diploid cells to repress haploid-specific gene expression Transcriptional -

Discovery of High Affinity Receptors for Dityrosine Through Inverse Virtual Screening and Docking and Molecular Dynamics
Article Discovery of High Affinity Receptors for Dityrosine through Inverse Virtual Screening and Docking and Molecular Dynamics Fangfang Wang 1,*,†, Wei Yang 2,3,† and Xiaojun Hu 1,* 1 School of Life Science, Linyi University, Linyi 276000, China; [email protected] 2 Department of Microbiology, Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia, [email protected] 3 Arieh Warshel Institute of Computational Biology, the Chinese University of Hong Kong, 2001 Longxiang Road, Longgang District, Shenzhen 518000, China * Corresponding author: [email protected] † These authors contributed equally to this work. Received: 09 December 2018; Accepted: 23 December 2018; Published: date Table S1. Docking affinity scores for cis-dityrosine binding to binding proteins. Target name PDB/UniProtKB Type Affinity (kcal/mol) Galectin-1 1A78/P56217 Lectin -6.2±0.0 Annexin III 1AXN/P12429 Calcium/phospholipid Binding Protein -7.5±0.0 Calmodulin 1CTR/P62158 Calcium Binding Protein -5.8±0.0 Seminal Plasma Protein Pdc-109 1H8P/P02784 Phosphorylcholine Binding Protein -6.6±0.0 Annexin V 1HAK/P08758 Calcium/phospholipid Binding -7.4±0.0 Alpha 1 antitrypsin 1HP7/P01009 Protein Binding -7.6±0.0 Histidine-Binding Protein 1HSL/P0AEU0 Binding Protein -6.3±0.0 Intestinal Fatty Acid Binding Protein 1ICN/P02693 Binding Protein(fatty Acid) -9.1±0.0* Migration Inhibitory Factor-Related Protein 14 1IRJ/P06702 Metal Binding Protein -7.0±0.0 Lysine-, Arginine-, Ornithine-Binding Protein 1LST/P02911 Amino Acid Binding Protein -6.5±0.0 -

Inositol+ Inositol- YCR034W FEN1 Fatty Acid Elongase, Involved In
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by RERO DOC Digital Library Fold change VPA+/VPA- Systemic nameGene name Description Inositol+ Inositol- Fatty acid elongase, involved in sphingolipid biosynthesis; acts on fatty acids of up to 24 carbons YCR034W FEN1 in length; mutations have regulatory effects on 1,3- 2.46 2.86 Elongase, involved in fatty acid and sphingolipid biosynthesis; synthesizes very long chain 20-26- YLR372W SUR4 carbon fatty acids from C18-CoA primers; involved in 2.77 2.47 Putative transmembrane protein involved in the biotin biosynthesis pathway; responsible for uptake YNR056C BIO5 of 7-keto 8-aminopelargonic acid; BIO5 is in a cluster − -8.92 Low-affinity amino acid permease with broad substrate range, involved in uptake of asparagine, − YCL025C AGP1 glutamine, and other amino acids; expression is -3.77 General amino acid permease; localization to the − YKR039W GAP1 plasma membrane is regulated by nitrogen source -2.85 High-affinity leucine permease, functions as a YBR068C BAP2 branched-chain amino acid permease involved in the − -6.22 Dicarboxylic amino acid permease, mediates high- YPL265W DIP5 affinity and high-capacity transport of L-glutamate -2.22 -2.36 Permease that serves as a gamma-aminobutyrate (GABA) transport protein involved in the utilization of YDL210W UGA4 GABA as a nitrogen source; catalyzes the transport of − -2.26 Plasma membrane arginine permease, requires YEL063C CAN1 phosphatidyl ethanolamine (PE) for localization, − -2.31 High-affinity S-adenosylmethionine -

Genome Wide Analysis Identifies Sphingolipid Metabolism As a New Target of Valproic Acid" (2016)
Wayne State University Wayne State University Dissertations 1-1-2016 Genome Wide Analysis Identifies phinS golipid Metabolism As A New Target Of Valproic Acid Shyamalagauri Jadhav Jadhav Wayne State University, Follow this and additional works at: https://digitalcommons.wayne.edu/oa_dissertations Part of the Biology Commons, and the Molecular Biology Commons Recommended Citation Jadhav, Shyamalagauri Jadhav, "Genome Wide Analysis Identifies Sphingolipid Metabolism As A New Target Of Valproic Acid" (2016). Wayne State University Dissertations. 1545. https://digitalcommons.wayne.edu/oa_dissertations/1545 This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. GENOME WIDE ANALYSIS IDENTIFIES SPHINGOLIPID METABOLISM AS A NEW TARGET OF VALPROIC ACID by SHYAMALAGAURI JADHAV DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan In partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2016 MAJOR: BIOLOGICAL SCIENCES Approved By: _____________________________ Advisor Date _____________________________ _____________________________ _____________________________ ©COPYRIGHT BY SHYAMALAGAURI JADHAV 2016 All Rights Reserved DEDICATION I would like to dedicate my dissertation to my late mother Chandrakala Jadhav who was a constant support and inspiration through the years. ii ACKNOWLEDGEMENTS First and foremost I would like to thank my advisor, Dr. Miriam L. Greenberg for her intellectual, moral, financial, and emotional support throughout my graduate journey. The realization of this thesis would have been impossible without her constant encouragement and strong support. I would like to thank her for the confidence she had in my ability to think and design the project through; and for her guidance when needed. -

Organizing Multi-Enzyme Systems Into Programmable Materials for Biocatalysis
catalysts Review Organizing Multi-Enzyme Systems into Programmable Materials for Biocatalysis Min-Ju Seo and Claudia Schmidt-Dannert * Department of Biochemistry, Molecular Biology & Biophysics, University of Minnesota, Saint Paul, MN 55108, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-612-625-5782 Abstract: Significant advances in enzyme discovery, protein and reaction engineering have trans- formed biocatalysis into a viable technology for the industrial scale manufacturing of chemicals. Multi-enzyme catalysis has emerged as a new frontier for the synthesis of complex chemicals. How- ever, the in vitro operation of multiple enzymes simultaneously in one vessel poses challenges that require new strategies for increasing the operational performance of enzymatic cascade reactions. Chief among those strategies is enzyme co-immobilization. This review will explore how advances in synthetic biology and protein engineering have led to bioinspired co-localization strategies for the scaffolding and compartmentalization of enzymes. Emphasis will be placed on genetically en- coded co-localization mechanisms as platforms for future autonomously self-organizing biocatalytic systems. Such genetically programmable systems could be produced by cell factories or emerging cell-free systems. Challenges and opportunities towards self-assembling, multifunctional biocatalytic materials will be discussed. Keywords: biocatalysis; multi-enzyme; cascade reaction; biomanufacturing; biomaterials; immobi- Citation: Seo, M.-J.; lization; cell-free -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA