A Tutorial for Understanding Chemical Reactivity Through the Valence Bond Approach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Interstellar Formamide (NH2CHO), a Key Prebiotic Precursor
Interstellar formamide (NH2CHO), a key prebiotic precursor Ana López-Sepulcre,∗,y,z Nadia Balucani,{ Cecilia Ceccarelli,y Claudio Codella,x,y François Dulieu,k and Patrice Theulé? yUniv. Grenoble Alpes, IPAG, F-38000 Grenoble, France zInstitut de Radioastronomie Millimétrique (IRAM), 300 rue de la piscine, F-38406 Saint-Martin d’Hères, France {Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, I-06123 Perugia, Italy xINAF, Osservatorio Astrofisico di Arcetri, Largo E. Fermi 5, I-50125, Firenze, Italy kUniversité de Cergy-Pontoise, Observatoire de Paris, PSL University, Sorbonne Université, CNRS, LERMA, F-95000, Cergy-Pontoise, France ?Aix-Marseille Université, PIIM UMR-CNRS 7345, 13397 Marseille, France E-mail: [email protected] Abstract Formamide (NH2CHO) has been identified as a potential precursor of a wide variety arXiv:1909.11770v1 [astro-ph.SR] 25 Sep 2019 of organic compounds essential to life, and many biochemical studies propose it likely played a crucial role in the context of the origin of life on our planet. The detection of formamide in comets, which are believed to have –at least partially– inherited their current chemical composition during the birth of the Solar System, raises the question whether a non-negligible amount of formamide may have been exogenously delivered onto a very young Earth about four billion years ago. A crucial part of the effort to 1 answer this question involves searching for formamide in regions where stars and planets are forming today in our Galaxy, as this can shed light on its formation, survival, and chemical re-processing along the different evolutionary phases leading to a star and planetary system like our own. -

Systematic Approach for Chemical Reactivity
SYSTEMATIC APPROACH FOR CHEMICAL REACTIVITY EVALUATION A Dissertation by ABDULREHMAN AHMED ALDEEB Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY December 2003 Major Subject: Chemical Engineering SYSTEMATIC APPROACH FOR CHEMICAL REACTIVITY EVALUATION A Dissertation by ABDULREHMAN AHMED ALDEEB Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Approved as to style and content by: ____________________________ ____________________________ M. Sam Mannan Kenneth R. Hall (Chair of Committee) (Member) ____________________________ ____________________________ Mark T. Holtzapple Jerald A. Caton (Member) (Member) ____________________________ Kenneth R. Hall (Head of Department) December 2003 Major Subject: Chemical Engineering iii ABSTRACT Systematic Approach for Chemical Reactivity Evaluation. (December 2003) Abdulrehman Ahmed Aldeeb, B.S., Jordan University of Science & Technology; M.S., The University of Texas at Arlington Chair of Advisory Committee: Dr. M. Sam Mannan Under certain conditions, reactive chemicals may proceed into uncontrolled chemical reaction pathways with rapid and significant increases in temperature, pressure, and/or gas evolution. Reactive chemicals have been involved in many industrial incidents, and have harmed people, property, and the environment. Evaluation of reactive chemical hazards is critical to design and operate safer chemical plant processes. Much effort is needed for experimental techniques, mainly calorimetric analysis, to measure thermal reactivity of chemical systems. Studying all the various reaction pathways experimentally however is very expensive and time consuming. Therefore, it is essential to employ simplified screening tools and other methods to reduce the number of experiments and to identify the most energetic pathways. -

Chapter 6 – the Structure and Reactivity of Molecules the Structure of Molecules This Is the Golden Age of Instrumentation
Chapter 6 – The Structure and Reactivity of Molecules The Structure of Molecules This is the golden age of instrumentation. Many of the techniques you used in organic lab were used a hundred years ago as the sole method of structure determination. Remember the nucleus has been known for just over 100 years! Beginning in the 1920's and 30's through today, we have seen the invention and the development of many analytical techniques used in structure determination. Infrared, NMR, UV-visible (electronic), and ESR spectroscopies, electrochemistry, X-ray crystallography are the major ones. Sixty years ago, an X-ray crystal structure could be an entire Ph.D. dissertation. Today an undergraduate can frequently solve the same structure in a single afternoon. We will talk about some of these methods later in the chapter. Valence Shell Electron Pair Repulsion (VSEPR) Theory While determining molecular structure is increasingly simple, a simple method of predicting structure is still invaluable. The method of choice is VSEPR theory. You saw this in CHM 211 so we'll only briefly review it. This method is a little different from the book’s and can be found in my general chemistry notes. Movable images of the different shapes can be found at http://science.marshall.edu/castella/chm211/vsepr.html (You will need Java® to view this page. Also you will have to use the Java® control panel to add this site to the secure list.). 1) Draw the Lewis structure of the molecule. 2) Determine the base shape of the molecule by adding the number of atoms bound to the central atom to the number of lone pairs on the central atom. -

Interpreting the Periodic Table – Teacher Page
Name: _____________________ Period: ____ Interpreting the Periodic Table – Teacher Page TEKS 8.5 Matter and energy. The student knows that matter is composed of atoms and has chemical and physical properties. The student is expected to: (C) interpret the arrangement of the Periodic Table, including groups and periods, to explain how properties are used to classify elements; Pre-Teach This is an activity designed to help students demonstrate an understanding of the arrangement of the periodic table. Students should be familiar with how the periodic table is arranged by atomic number, mass, energy levels, groups, periods, reactivity, valence electrons, reactivity etc. through other forms of presentation before completing this activity. A good activity to help construct this information for students would be the Mr. X activity. Differentiation Having students work together on this activity can be beneficial in some instances, but can also hinder student’s recall of the information. This is intended to be a summative assessment for this particular TEK before moving on to another concept. It is recommended that the assignment be modified so that the patterns could be easier to identify by reducing the number of letters on the “hypothetical periodic table” and strategically placing them on the table so they are closer together. Name: _____________________ Period: ____ Interpreting the Periodic Table Examine the hypothetical periodic table shown below. Use this periodic table to answer the questions that follow. B A D E C G F I H 1. Which pair(s) of elements has the same number of valence electrons? _____________________________________________________________ 2. How many valence electrons do they have? _____________________________________________________________ 3. -

Trends in Reactivity in the Periodic Table
Chemistry for the gifted and talented 35 Trends in reactivity in the Periodic Table Student worksheet: CDROM index 18SW Discussion of answers: CDROM index 18DA Topics Trends in reactivity of Groups 1 and 7 and the ionic bonding model. Level Very able students aged 14–16. Prior knowledge Ionic bonding and the Periodic Table. Rationale This activity aims to: • help students develop a tool (flowcharts) to aid organisation of their line of reasoning; • help students explore links between trends in the reactivity of Groups 1 and 7 and atomic structure; • give students the opportunity to use and critically evaluate the relevance of ionisation energy data to the reactivity series of metals; •reinforce the idea of chemical changes being driven by energy changes; and • challenge the idea that Group 1 metals want to give away their outer shell electron. Use This could be used to follow up some work on the Periodic Table where the trends in reactivity in Groups 1 and 7 have been identified. It can be used as a differentiated activity for the more able students within a group. When the students have completed the worksheet they should be given the Discussion of answers sheet. They could check their own work or conduct a peer review of the work of Chemistry for the gifted and talented Trends in reactivity in the Periodic Table Flow charts are sometimes a good way to organise your thoughts into a line of reasoning or logical argument. The example below is about the question of why Group 1 elements get more reactive as you go down the group. -
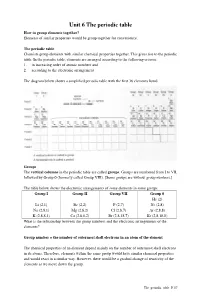
Unit 6 the Periodic Table How to Group Elements Together? Elements of Similar Properties Would Be Group Together for Convenience
Unit 6 The periodic table How to group elements together? Elements of similar properties would be group together for convenience. The periodic table Chemists group elements with similar chemical properties together. This gives rise to the periodic table. In the periodic table, elements are arranged according to the following criteria: 1. in increasing order of atomic numbers and 2. according to the electronic arrangement The diagram below shows a simplified periodic table with the first 36 elements listed. Groups The vertical columns in the periodic table are called groups . Groups are numbered from I to VII, followed by Group 0 (formerly called Group VIII). [Some groups are without group numbers.] The table below shows the electronic arrangements of some elements in some groups. Group I Group II Group VII Group 0 He (2) Li (2,1) Be (2,2) F (2,7) Ne (2,8) Na (2,8,1) Mg (2,8,2) Cl (2,8,7) Ar (2,8,8) K (2,8,8,1) Ca (2,8,8,2) Br (2,8,18,7) Kr (2,8,18,8) What is the relationship between the group numbers and the electronic arrangements of the elements? Group number = the number of outermost shell electrons in an atom of the element The chemical properties of an element depend mainly on the number of outermost shell electrons in its atoms. Therefore, elements within the same group would have similar chemical properties and would react in a similar way. However, there would be a gradual change of reactivity of the elements as we move down the group. -

Periodic Table of the Elements Notes
Periodic Table of the Elements Notes Arrangement of the known elements based on atomic number and chemical and physical properties. Divided into three basic categories: Metals (left side of the table) Nonmetals (right side of the table) Metalloids (touching the zig zag line) Basic Organization by: Atomic structure Atomic number Chemical and Physical Properties Uses of the Periodic Table Useful in predicting: chemical behavior of the elements trends properties of the elements Atomic Structure Review: Atoms are made of protons, electrons, and neutrons. Elements are atoms of only one type. Elements are identified by the atomic number (# of protons in nucleus). Energy Levels Review: Electrons are arranged in a region around the nucleus called an electron cloud. Energy levels are located within the cloud. At least 1 energy level and as many as 7 energy levels exist in atoms Energy Levels & Valence Electrons Energy levels hold a specific amount of electrons: 1st level = up to 2 2nd level = up to 8 3rd level = up to 8 (first 18 elements only) The electrons in the outermost level are called valence electrons. Determine reactivity - how elements will react with others to form compounds Outermost level does not usually fill completely with electrons Using the Table to Identify Valence Electrons Elements are grouped into vertical columns because they have similar properties. These are called groups or families. Groups are numbered 1-18. Group numbers can help you determine the number of valence electrons: Group 1 has 1 valence electron. Group 2 has 2 valence electrons. Groups 3–12 are transition metals and have 1 or 2 valence electrons. -

The Reactivity Properties of Hydrogen Storage Materials in the Context of Systems
Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy’s National Nuclear Security Administration under contract DE-AC04-94AL85000. The reactivity properties of hydrogen storage materials in the context of systems 2009 DOE H2 Program Annual Merit Review May 20th, 2009 Daniel E. Dedrick Sandia National Laboratories Mike Kanouff, Rich Larson, Bob Bradshaw, George Sartor, Richard Behrens, LeRoy Whinnery, Joe Cordaro, Aaron Highley stp_51_dedrick This presentation does not contain any proprietary, confidential, or otherwise restricted information Overview Timeline Barriers • Start: July 2007 • End: September 2010 On-Board Hydrogen Storage –Durability/Operability (D) • Percent complete: 60% –Codes and Standards (F) –Reproducibility of Performance (Q) Budget Partners • $2.1M SRNL - Anton UTRC – Mosher (100% DOE H program) 2 IPHE • 630K in FY08 • 750K for FY09 Relevance: Overall Objective Develop generalized methods and procedures required to quantify the effects of hydrogen storage material contamination in an automotive environment Eventual Impact: • Enable the design, handling and operation of effective hydrogen storage systems for automotive applications. • Provide technical basis for C&S efforts when appropriate technology maturity has been attained. Approach: Project organized into three inter- dependent and collaborative tasks Task 1 - Quantify fundamental processes and hazards of material contamination (SRNL, UTRC, IPHE) – Illuminates the fundamental contamination -

Reversing the Native Aerobic Oxidation Reactivity of Graphitic Carbon: Heterogeneous Metal-Free Alkene Hydrogenation
Reversing the Native Aerobic Oxidation Reactivity of Graphitic Carbon: Heterogeneous Metal-Free Alkene Hydrogenation The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Murray, Alexander T. and Yogesh Surendranath. “Reversing the Native Aerobic Oxidation Reactivity of Graphitic Carbon: Heterogeneous Metal-Free Alkene Hydrogenation.” ACS Catalysis 7, 5 (April 2017): 3307–3312 © 2017 American Chemical Society As Published http://dx.doi.org/10.1021/acscatal.7b00395 Publisher American Chemical Society (ACS) Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/115122 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Reversing The Native Aerobic Oxidation Reactivity Of Graphitic Car- bon: Heterogeneous Metal-Free Alkene Hydrogenation Alexander T. Murray and Yogesh Surendranath* Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States. KEYWORDS Hydrogenation, Carbon Black, Carbocatalysis, Diimide, Alkene ABSTRACT: Commercially available carbon blacks serve as effective metal-free catalysts for the selective hydrogenation of car- bon-carbon multiple bonds under aerobic conditions using hydrazine as the terminal reductant. The reaction, which proceeds through a putative diimide intermediate, displays high tolerance to a variety of functional groups, including those sensitive to nu- cleophilic displacement by hydrazine, aerobic oxidation, or hydrazine-mediated reduction. Hydrazine chemisorbs strongly to the carbon surface, attenuating its native oxidative reactivity and allowing for selective hydrogenation. The catalytic sequence estab- lished here effectively umpolungs the reactivity of carbon, thereby enabling the use of this low cost material in selective reduction catalysis. -

Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases
International Journal of Molecular Sciences Review Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases Fabrice Collin Laboratoire des IMRCP, Université de Toulouse, CNRS UMR 5623, Université Toulouse III-Paul Sabatier, 118 Route de Narbonne, 31062 Toulouse CEDEX 09, France; [email protected] Received: 26 April 2019; Accepted: 13 May 2019; Published: 15 May 2019 Abstract: Increasing numbers of individuals suffer from neurodegenerative diseases, which are characterized by progressive loss of neurons. Oxidative stress, in particular, the overproduction of Reactive Oxygen Species (ROS), play an important role in the development of these diseases, as evidenced by the detection of products of lipid, protein and DNA oxidation in vivo. Even if they participate in cell signaling and metabolism regulation, ROS are also formidable weapons against most of the biological materials because of their intrinsic nature. By nature too, neurons are particularly sensitive to oxidation because of their high polyunsaturated fatty acid content, weak antioxidant defense and high oxygen consumption. Thus, the overproduction of ROS in neurons appears as particularly deleterious and the mechanisms involved in oxidative degradation of biomolecules are numerous and complexes. This review highlights the production and regulation of ROS, their chemical properties, both from kinetic and thermodynamic points of view, the links between them, and their implication in neurodegenerative diseases. Keywords: reactive oxygen species; superoxide anion; hydroxyl radical; hydrogen peroxide; hydroperoxides; neurodegenerative diseases; NADPH oxidase; superoxide dismutase 1. Introduction Reactive Oxygen Species (ROS) are radical or molecular species whose physical-chemical properties are well-known both on thermodynamic and kinetic points of view. -

Conjugated, Carbon-Centered Radicals
molecules Review Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals Takashi Kubo Department of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan; [email protected] Received: 26 January 2019; Accepted: 11 February 2019; Published: 13 February 2019 Abstract: Recently, long-lived, organic radical species have attracted much attention from chemists and material scientists because of their unique electronic properties derived from their magnetic spin and singly occupied molecular orbitals. Most stable and persistent organic radicals are heteroatom-centered radicals, whereas carbon-centered radicals are generally very reactive and therefore have had limited applications. Because the physical properties of carbon-centered radicals depend predominantly on the topology of the π-electron array, the development of new carbon-centered radicals is key to new basic molecular skeletons that promise novel and diverse applications of spin materials. This account summarizes our recent studies on the development of novel carbon-centered radicals, including phenalenyl, fluorenyl, and triarylmethyl radicals. Keywords: π-conjugated radicals; hydrocarbon radicals; persistent; anthryl; phenalenyl; fluorenyl 1. Introduction Organic radical species are generally recognized as highly reactive, intermediate species. However, recently, functional materials taking advantage of the feature of open-shell electronic structure have attracted much attention from chemists and material scientists; therefore, the development of novel, long-lived, organic, radical species becomes more important [1–7]. Nitronyl nitroxides, galvinoxyl, and DPPH are well known as stable, organic, radical species, which are commercially available chemicals. These stable radical species are “heteroatom-centered radicals”, in which unpaired electrons are mainly distributed on heteroatoms. -

Periodic Table Key Concepts
Periodic Table Key Concepts Periodic Table Basics The periodic table is a table of all the elements which make up matter Elements initially grouped in a table by Dmitri Mendeleev Symbols – each element has a symbol which is either a Capital Letter or a Capital Letter followed by a lower case letter Atomic Number – the number above an element’s symbol which shows the number of protons Atomic Mass – the number found below an elements symbol which shows the mass of the element. Mass = the number of protons + the number of neutrons Metals – the elements which have the properties of malleability, luster, and conductivity o These elements are good conductors of electricity & heat. o Found to the left of the zig-zag line on the periodic table Nonmetals – do not have the properties of metals. Found to the right of the zig-zag line Metalloids – elements found along the zig-zag line of the periodic table and have some properties of metals and nonmetals (B, Si, Ge, As, Sb, Te, and Po) Groups The columns going up and down (There are 18 groups) Group 1: Hydrogen, Lithium, Sodium, Potassium, Rubidium, Cesium, and Francium Elements arranged so that elements with similar properties would be in the same group. o Group 1 Alkali Metals - highly reactive metals o Group 2 Alkali Earth Metals – reactive metals o Group 3-12 Transition Metals o Group 17 Halogens – highly reactive non-metals o Group 18 Noble Gases - do not react or combine with any other elements. Elements are grouped according to their properties or reactivity Reactivity is determined by the number of electrons in an element’s outer energy level These electrons are called valence electrons Periods The rows that run from left to right on the periodic table (There are 7 periods) Period 1 contains 2 elements, Hydrogen and Helium.