Pharmaceutical Compositions and Dosage Forms of Thalidomide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
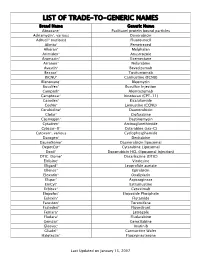
Trade-To-Generic Names
LIST OF TRADE-TO-GENERIC NAMES Brand Name Generic Name Abraxane® Paclitaxel protein bound particles Adriamycin®, various Doxorubicin Adrucil® (various) Fluorouracil Alimta® Pemetrexed Alkeran® Melphalan Arimidex® Anastrozole Aromasin® Exemestane Arranon® Nelarabine Avastin® Bevacizumab Bexxar® Tositumomab BiCNU® Carmustine (BCNU) Blenoxane® Bleomycin Busulfex® Busulfan Injection Campath® Alemtuzumab Camptosar® Irinotecan (CPT-11) Casodex® Bicalutamide CeeNu® Lomustine (CCNU) Cerubidine® Daunorubicin Clolar® Clofarabine Cosmegen® Dactinomycin Cytadren® Aminoglutethimide Cytosar-U® Cytarabine (ara-C) Cytoxan®, various Cyclophosphamide Dacogen® Decitabine DaunoXome® Daunorubicin liposomal DepotCyt® Cytarabine Liposomal Doxil® Doxorubicin HCL (liposomal injection) DTIC-Dome® Dacarbazine (DTIC) Eldisine® Vindesine Eligard® Leuprolide acetate Ellence® Epirubicin Eloxatin® Oxaliplatin Elspar® Asparaginase EmCyt® Estramustine Erbitux® Cetuximab Etopofos® Etoposide Phosphate Eulexin® Flutamide Fareston® Toremifene Faslodex® Fluvestrant Femara® Letrozole Fludara® Fludarabine Gemzar® Gemcitabine Gleevec® Imatinib Gliadel® Carmustine Wafer Halotestin® Fluoxymesterone Last Updated on January 15, 2007 Brand Name Generic Name Herceptin® Trastuzumab Hexalen® Altretamine Hycamtin® Topotecan Hydrea® Hydroxyurea Idamycin® Idarubicin Ifex® Ifosfamide Intron A® Interferon alfa-2b Iressa® Gefitinib Leukeran® Chlorambucil Leukine® Sargramostim Leustatin® Cladribine Lupron depot® Leuprolide acetate depot Lupron® Leuprolide acetate Matulane® Procarbazine Megace® -

Transcatheter Arterial Chemoembolization Therapy for Hepatocellular Carcinoma Using Polylactic Acid Microspheres Containing Acla
[CANCER RESEARCH 49. 4357-4362, August 1. 1989] Transcatheter Arterial Chemoembolization Therapy for Hepatocellular Carcinoma Using Polylactic Acid Microspheres Containing Aclarubicin Hydrochloride 1omonimi Ichihara,1 Kiyoshi Sakamoto, Katsutaka Mori, and Masanobu Akagi Department of Surgery II, Kumamoto University Medical School, Kumamoto 860, Japan 4BSTRACT MATERIALS AND METHODS Transcatheter arterial Chemoembolization therapy using polylactic Preparation of PLA-ACRms acid microspheres containing aclarubicin hydrochloride (ACR) was per PLA-ACRms were prepared in the pharmacy of the Kumamoto formed in 62 patients with primary hepatocellular carcinoma. These microspheres were about 200 ¡anin diameter and contained 10% (w/w) University Hospital. Briefly, aclarubicin hydrochloride and isopropyl aclarubicin. A single dose of polylactic acid microspheres containing ACR myristate, a medium-chain fatty acid ester, were dissolved in 7.5% (50-100 mg of ACR) was administered 1 to 8 times with a mean of 2.2 polylactic acid-methylene chloride. The resultant solution was dispersed in 1% gelatin solution and sterilized in an autoclave for 20 min at doses (a total of 160 treatments) in 62 patients. Antitumor effects were 120°C;thismixture was then stirred with a magnetic stirrer at 500 rpm observed from the decrease in serum a-fetoprotein levels (82.1% of the patients) and in two dimensional size of tumor on computed tomography for 1 h. The resultant microspheres were collected by filtration through (93.6%). The cumulative survival rate was 54.3% at 1 year, 24.6% at 2 a membrane filter (3 //m pore diameter), rinsed 2 to 3 times (in 1 liter years, and 19.2% at 3 years, respectively, among 59 patients with of distilled water), and dried under reduced pressure for 5 days. -

Proteomics and Drug Repurposing in CLL Towards Precision Medicine
cancers Review Proteomics and Drug Repurposing in CLL towards Precision Medicine Dimitra Mavridou 1,2,3, Konstantina Psatha 1,2,3,4,* and Michalis Aivaliotis 1,2,3,4,* 1 Laboratory of Biochemistry, School of Medicine, Faculty of Health Sciences, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece; [email protected] 2 Functional Proteomics and Systems Biology (FunPATh)—Center for Interdisciplinary Research and Innovation (CIRI-AUTH), GR-57001 Thessaloniki, Greece 3 Basic and Translational Research Unit, Special Unit for Biomedical Research and Education, School of Medicine, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece 4 Institute of Molecular Biology and Biotechnology, Foundation of Research and Technology, GR-70013 Heraklion, Greece * Correspondence: [email protected] (K.P.); [email protected] (M.A.) Simple Summary: Despite continued efforts, the current status of knowledge in CLL molecular pathobiology, diagnosis, prognosis and treatment remains elusive and imprecise. Proteomics ap- proaches combined with advanced bioinformatics and drug repurposing promise to shed light on the complex proteome heterogeneity of CLL patients and mitigate, improve, or even eliminate the knowledge stagnation. In relation to this concept, this review presents a brief overview of all the available proteomics and drug repurposing studies in CLL and suggests the way such studies can be exploited to find effective therapeutic options combined with drug repurposing strategies to adopt and accost a more “precision medicine” spectrum. Citation: Mavridou, D.; Psatha, K.; Abstract: CLL is a hematological malignancy considered as the most frequent lymphoproliferative Aivaliotis, M. Proteomics and Drug disease in the western world. It is characterized by high molecular heterogeneity and despite the Repurposing in CLL towards available therapeutic options, there are many patient subgroups showing the insufficient effectiveness Precision Medicine. -

A Phase II Study of Paclitaxel and Capecitabine As a First-Line Combination Chemotherapy for Advanced Gastric Cancer
British Journal of Cancer (2008) 98, 316 – 322 & 2008 Cancer Research UK All rights reserved 0007 – 0920/08 $30.00 www.bjcancer.com A phase II study of paclitaxel and capecitabine as a first-line combination chemotherapy for advanced gastric cancer Clinical Studies HJ Kang1, HM Chang1, TW Kim1, M-H Ryu1, H-J Sohn1, JH Yook2,STOh2, BS Kim2, J-S Lee1 and Y-K Kang*,1 1 2 Division of Oncology, Department of Medicine, University of Ulsan College of Medicine, Asan Medical Center, Seoul, Korea; Department of Surgery, University of Ulsan College of Medicine, Asan Medical Center, Seoul, Korea Paclitaxel and capecitabine, which have distinct mechanisms of action and toxicity profiles, have each shown high activity as single agents in gastric cancer. Synergistic interaction between these two drugs was suggested by taxane-induced upregulation of thymidine phosphorylase. We, therefore, evaluated the antitumour activity and toxicities of paclitaxel and capecitabine as first-line therapy in patients with advanced gastric cancer (AGC). Patients with histologically confirmed unresectable or metastatic AGC were treated À2 À2 with capecitabine 825 mg m p.o. twice daily on days 1–14 and paclitaxel 175 mg m i.v. on day 1 every 3 weeks until disease progression or unacceptable toxicities. Between June 2002 and May 2004, 45 patients, of median age 57 years (range ¼ 38–73 years), were treated with the combination of capecitabine and paclitaxel. After a median 6 cycles (range ¼ 1–9 cycles) of chemotherapy, 43 were evaluable for toxicity and response. A total of 2 patients showed complete response and 20 showed partial response making the overall response rate 48.9% (95% CI ¼ 30.3–63.5%). -

Us 8530498 B1 3
USOO853 0498B1 (12) UnitedO States Patent (10) Patent No.: US 8,530,498 B1 Zeldis (45) Date of Patent: *Sep. 10, 2013 (54) METHODS FORTREATING MULTIPLE 5,639,476 A 6/1997 OShlack et al. MYELOMAWITH 5,674,533 A 10, 1997 Santus et al. 3-(4-AMINO-1-OXO-1,3-DIHYDROISOINDOL- 395 A 22 N. 2-YL)PIPERIDINE-2,6-DIONE 5,731,325 A 3/1998 Andrulis, Jr. et al. 5,733,566 A 3, 1998 Lewis (71) Applicant: Celgene Corporation, Summit, NJ (US) 5,798.368 A 8, 1998 Muller et al. 5,874.448 A 2f1999 Muller et al. (72) Inventor: Jerome B. Zeldis, Princeton, NJ (US) 5,877,200 A 3, 1999 Muller 5,929,117 A 7/1999 Muller et al. 5,955,476 A 9, 1999 Muller et al. (73) Assignee: Celgene Corporation, Summit, NJ (US) 6,020,358 A 2/2000 Muller et al. - 6,071,948 A 6/2000 D'Amato (*) Notice: Subject to any disclaimer, the term of this 6,114,355 A 9, 2000 D'Amato patent is extended or adjusted under 35 SS f 1939. All et al. U.S.C. 154(b) by 0 days. 6,235,756 B1 5/2001 D'Amatoreen et al. This patent is Subject to a terminal dis- 6,281.230 B1 8/2001 Muller et al. claimer 6,316,471 B1 1 1/2001 Muller et al. 6,326,388 B1 12/2001 Man et al. 6,335,349 B1 1/2002 Muller et al. (21) Appl. No.: 13/858,708 6,380.239 B1 4/2002 Muller et al. -

Multinational Evaluation of Mycophenolic Acid, Tacrolimus
View metadata, citation and similar papers at core.ac.uk brought to you by CORE providedORIGINAL by University of QueenslandPAPER eSpace ISSN 1425-9524 © Ann Transplant, 2016; 21: 1-11 DOI: 10.12659/AOT.895664 Received: 2015.08.15 Accepted: 2015.09.01 Multinational Evaluation of Mycophenolic Published: 2016.01.05 Acid, Tacrolimus, Cyclosporin, Sirolimus, and Everolimus Utilization Authors’ Contribution: ABCDEF Kyle M. Gardiner School of Pharmacy, University of Queensland, Brisbane, QLD, Australia Study Design A ACDEF Susan E. Tett Data Collection B Statistical Analysis C ACDEF Christine E. Staatz Data Interpretation D Manuscript Preparation E Literature Search F Funds Collection G Corresponding Author: Christine E. Staatz, e-mail: [email protected] Source of support: Departmental funding only Background: Increasing immunosuppressant utilization and expenditure is a worldwide challenge as more people success- fully live with transplanted organs. Our aims were to characterize utilization of mycophenolate, tacrolimus, cy- closporin, sirolimus, and everolimus in Australian transplant recipients from 2007 to 2013; to identify specific patterns of usage; and to compare Australian utilization with Norwegian, Danish, Swedish, and the Netherlands use. Material/Methods: Australian utilization and expenditure data were captured through national Pharmaceutical Benefits Scheme and Highly Specialized Drug administrative databases. Norwegian, Danish, Swedish, and the Netherlands uti- lization were retrieved from their healthcare databases. Utilization was compared as defined daily dose per 1000 population per day (DDD/1000 population/day). Data on kidney transplant recipients, the predominant patient group prescribed these medicines, were obtained from international transplant registries. Results: From 2007–2013 Australian utilization of mycophenolic acid, tacrolimus and everolimus increased 2.7-fold, 2.2- fold, and 2.3-fold, respectively. -

Phenotype Microarrays Panels PM-M1 to PM-M14
Phenotype MicroArrays™ Panels PM-M1 to PM-M14 for Phenotypic Characterization of Mammalian Cells Assays: Energy Metabolism Pathways Ion and Hormone Effects on Cells Sensitivity to Anti-Cancer Agents and for Optimizing Culture Conditions for Mammalian Cells PRODUCT DESCRIPTIONS AND INSTRUCTIONS FOR USE PM-M1 Cat. #13101 PM-M2 Cat. #13102 PM-M3 Cat. #13103 PM-M4 Cat. #13104 PM-M5 Cat. #13105 PM-M6 Cat. #13106 PM-M7 Cat. #13107 PM-M8 Cat. #13108 PM-M11 Cat. #13111 PM-M12 Cat. #13112 PM-M13 Cat. #13113 PM-M14 Cat. #13114 © 2016 Biolog, Inc. All rights reserved Printed in the United States of America 00P 134 Rev F February 2020 - 1 - CONTENTS I. Introduction ...................................................................................................... 2 a. Overview ................................................................................................... 2 b. Background ............................................................................................... 2 c. Uses ........................................................................................................... 2 d. Advantages ................................................................................................ 3 II. Product Description, PM-M1 to M4 ................................................................ 3 III. Protocols, PM-M1 to M4 ................................................................................. 7 a. Materials Required .................................................................................... 7 b. Determination -

Classification Decisions Taken by the Harmonized System Committee from the 47Th to 60Th Sessions (2011
CLASSIFICATION DECISIONS TAKEN BY THE HARMONIZED SYSTEM COMMITTEE FROM THE 47TH TO 60TH SESSIONS (2011 - 2018) WORLD CUSTOMS ORGANIZATION Rue du Marché 30 B-1210 Brussels Belgium November 2011 Copyright © 2011 World Customs Organization. All rights reserved. Requests and inquiries concerning translation, reproduction and adaptation rights should be addressed to [email protected]. D/2011/0448/25 The following list contains the classification decisions (other than those subject to a reservation) taken by the Harmonized System Committee ( 47th Session – March 2011) on specific products, together with their related Harmonized System code numbers and, in certain cases, the classification rationale. Advice Parties seeking to import or export merchandise covered by a decision are advised to verify the implementation of the decision by the importing or exporting country, as the case may be. HS codes Classification No Product description Classification considered rationale 1. Preparation, in the form of a powder, consisting of 92 % sugar, 6 % 2106.90 GRIs 1 and 6 black currant powder, anticaking agent, citric acid and black currant flavouring, put up for retail sale in 32-gram sachets, intended to be consumed as a beverage after mixing with hot water. 2. Vanutide cridificar (INN List 100). 3002.20 3. Certain INN products. Chapters 28, 29 (See “INN List 101” at the end of this publication.) and 30 4. Certain INN products. Chapters 13, 29 (See “INN List 102” at the end of this publication.) and 30 5. Certain INN products. Chapters 28, 29, (See “INN List 103” at the end of this publication.) 30, 35 and 39 6. Re-classification of INN products. -

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

WO 2017/173206 Al 5 October 2017 (05.10.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2017/173206 Al 5 October 2017 (05.10.2017) P O P C T (51) International Patent Classification: CA 94121 (US). HUBBARD, Robert; 7684 Marker Road, A61K 31/52 (2006.01) C07D 473/02 (2006.01) San Diego, CA 92087 (US). MIKOLON, David; 6140 A61K 31/505 (2006.01) C07D 473/26 (2006.01) Calle Empinada, San Diego, CA 92120 (US). RAYMON, A61K 31/519 (2006.01) C07D 473/32 (2006.01) Heather; 3520 Vista de la Orilla, San Diego, CA 921 17 (US). SHI, Tao; 4650 Tarantella Lane, San Diego, CA (21) International Application Number: 92130 (US). TRAN, Tam, M.; 8953 Libra Drive, San PCT/US20 17/025252 Diego, CA 92126 (US). TSUJI, Toshiya; 4171 Donald (22) International Filing Date: Court, San Diego, CA 921 17 (US). WONG, Lilly, L.; 871 3 1 March 2017 (3 1.03.2017) Viva Court, Solana Beach, CA 92075 (US). XU, Suichan; 9650 Deer Trail Place, San Diego, CA 92127 (US). ZHU, (25) Filing Language: English Dan; 4432 Calle Mar De Armonia, San Diego, CA 92130 (26) Publication Language: English (US). (30) Priority Data: (74) Agents: BRUNER, Michael, J. et al; Jones Day, 250 Ve- 62/3 17,412 1 April 2016 (01.04.2016) US sey Street, New York, NY 10281-1047 (US). (71) Applicant: SIGNAL PHARMACEUTICALS, LLC (81) Designated States (unless otherwise indicated, for every [US/US]; 10300 Campus Point Drive, Suite 100, San kind of national protection available): AE, AG, AL, AM, Diego, CA 92121 (US). -

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol