This Item Is the Archived Peer-Reviewed Author-Version Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(Dendrocopos Syriacus) and Middle Spotted Woodpecker (Dendrocoptes Medius) Around Yasouj City in Southwestern Iran
Archive of SID Iranian Journal of Animal Biosystematics (IJAB) Vol.15, No.1, 99-105, 2019 ISSN: 1735-434X (print); 2423-4222 (online) DOI: 10.22067/ijab.v15i1.81230 Comparison of nest holes between Syrian Woodpecker (Dendrocopos syriacus) and Middle Spotted Woodpecker (Dendrocoptes medius) around Yasouj city in Southwestern Iran Mohamadian, F., Shafaeipour, A.* and Fathinia, B. Department of Biology, Faculty of Science, Yasouj University, Yasouj, Iran (Received: 10 May 2019; Accepted: 9 October 2019) In this study, the nest-cavity characteristics of Middle Spotted and Syrian Woodpeckers as well as tree characteristics (i.e. tree diameter at breast height and hole measurements) chosen by each species were analyzed. Our results show that vertical entrance diameter, chamber vertical depth, chamber horizontal depth, area of entrance and cavity volume were significantly different between Syrian Woodpecker and Middle Spotted Woodpecker (P < 0.05). The average tree diameter at breast height and nest height between the two species was not significantly different (P > 0.05). For both species, the tree diameter at breast height and nest height did not significantly correlate. The directions of nests’ entrances were different in the two species, not showing a preferentially selected direction. These two species chose different habitats with different tree coverings, which can reduce the competition between the two species over selecting a tree for hole excavation. Key words: competition, dimensions, nest-cavity, primary hole nesters. INTRODUCTION Woodpeckers (Picidae) are considered as important excavator species by providing cavities and holes to many other hole-nesting species (Cockle et al., 2011). An important part of habitat selection in bird species is where to choose a suitable nest-site (Hilden, 1965; Stauffer & Best, 1982; Cody, 1985). -
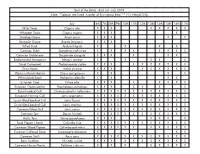
Best of the Baltic - Bird List - July 2019 Note: *Species Are Listed in Order of First Seeing Them ** H = Heard Only
Best of the Baltic - Bird List - July 2019 Note: *Species are listed in order of first seeing them ** H = Heard Only July 6th 7th 8th 9th 10th 11th 12th 13th 14th 15th 16th 17th Mute Swan Cygnus olor X X X X X X X X Whopper Swan Cygnus cygnus X X X X Greylag Goose Anser anser X X X X X Barnacle Goose Branta leucopsis X X X Tufted Duck Aythya fuligula X X X X Common Eider Somateria mollissima X X X X X X X X Common Goldeneye Bucephala clangula X X X X X X Red-breasted Merganser Mergus serrator X X X X X Great Cormorant Phalacrocorax carbo X X X X X X X X X X Grey Heron Ardea cinerea X X X X X X X X X Western Marsh Harrier Circus aeruginosus X X X X White-tailed Eagle Haliaeetus albicilla X X X X Eurasian Coot Fulica atra X X X X X X X X Eurasian Oystercatcher Haematopus ostralegus X X X X X X X Black-headed Gull Chroicocephalus ridibundus X X X X X X X X X X X X European Herring Gull Larus argentatus X X X X X X X X X X X X Lesser Black-backed Gull Larus fuscus X X X X X X X X X X X X Great Black-backed Gull Larus marinus X X X X X X X X X X X X Common/Mew Gull Larus canus X X X X X X X X X X X X Common Tern Sterna hirundo X X X X X X X X X X X X Arctic Tern Sterna paradisaea X X X X X X X Feral Pigeon ( Rock) Columba livia X X X X X X X X X X X X Common Wood Pigeon Columba palumbus X X X X X X X X X X X Eurasian Collared Dove Streptopelia decaocto X X X Common Swift Apus apus X X X X X X X X X X X X Barn Swallow Hirundo rustica X X X X X X X X X X X Common House Martin Delichon urbicum X X X X X X X X White Wagtail Motacilla alba X X -
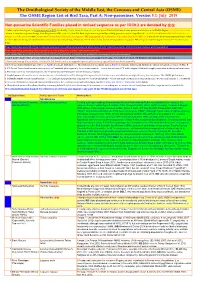
ORL 5.1 Non-Passerines Final Draft01a.Xlsx
The Ornithological Society of the Middle East, the Caucasus and Central Asia (OSME) The OSME Region List of Bird Taxa, Part A: Non-passerines. Version 5.1: July 2019 Non-passerine Scientific Families placed in revised sequence as per IOC9.2 are denoted by ֍֍ A fuller explanation is given in Explanation of the ORL, but briefly, Bright green shading of a row (eg Syrian Ostrich) indicates former presence of a taxon in the OSME Region. Light gold shading in column A indicates sequence change from the previous ORL issue. For taxa that have unproven and probably unlikely presence, see the Hypothetical List. Red font indicates added information since the previous ORL version or the Conservation Threat Status (Critically Endangered = CE, Endangered = E, Vulnerable = V and Data Deficient = DD only). Not all synonyms have been examined. Serial numbers (SN) are merely an administrative convenience and may change. Please do not cite them in any formal correspondence or papers. NB: Compass cardinals (eg N = north, SE = southeast) are used. Rows shaded thus and with yellow text denote summaries of problem taxon groups in which some closely-related taxa may be of indeterminate status or are being studied. Rows shaded thus and with yellow text indicate recent or data-driven major conservation concerns. Rows shaded thus and with white text contain additional explanatory information on problem taxon groups as and when necessary. English names shaded thus are taxa on BirdLife Tracking Database, http://seabirdtracking.org/mapper/index.php. Nos tracked are small. NB BirdLife still lump many seabird taxa. A broad dark orange line, as below, indicates the last taxon in a new or suggested species split, or where sspp are best considered separately. -

Pdf 184.33 K
Iranian Journal of Animal Biosystematics (IJAB) Vol.15, No.1, 99-105, 2019 ISSN: 1735-434X (print); 2423-4222 (online) DOI: 10.22067/ijab.v15i1.81230 Comparison of nest holes between Syrian Woodpecker ( Dendrocopos syriacus ) and Middle Spotted Woodpecker ( Dendrocoptes medius ) around Yasouj city in Southwestern Iran Mohamadian, F., Shafaeipour, A.* and Fathinia, B. Department of Biology, Faculty of Science, Yasouj University, Yasouj, Iran (Received: 10 May 2019; Accepted: 9 October 2019) In this study, the nest-cavity characteristics of Middle Spotted and Syrian Woodpeckers as well as tree characteristics (i.e. tree diameter at breast height and hole measurements) chosen by each species were analyzed. Our results show that vertical entrance diameter, chamber vertical depth, chamber horizontal depth, area of entrance and cavity volume were significantly different between Syrian Woodpecker and Middle Spotted Woodpecker (P < 0.05). The average tree diameter at breast height and nest height between the two species was not significantly different (P > 0.05). For both species, the tree diameter at breast height and nest height did not significantly correlate. The directions of nests’ entrances were different in the two species, not showing a preferentially selected direction. These two species chose different habitats with different tree coverings, which can reduce the competition between the two species over selecting a tree for hole excavation. Key words: competition, dimensions, nest-cavity, primary hole nesters. INTRODUCTION Woodpeckers (Picidae) are considered as important excavator species by providing cavities and holes to many other hole-nesting species (Cockle et al ., 2011). An important part of habitat selection in bird species is where to choose a suitable nest-site (Hilden, 1965; Stauffer & Best, 1982; Cody, 1985). -
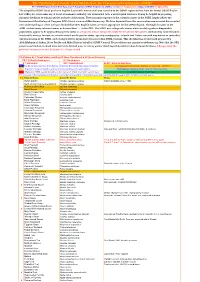
Simplified-ORL-2019-5.1-Final.Pdf
The Ornithological Society of the Middle East, the Caucasus and Central Asia (OSME) The OSME Region List of Bird Taxa, Part F: Simplified OSME Region List (SORL) version 5.1 August 2019. (Aligns with ORL 5.1 July 2019) The simplified OSME list of preferred English & scientific names of all taxa recorded in the OSME region derives from the formal OSME Region List (ORL); see www.osme.org. It is not a taxonomic authority, but is intended to be a useful quick reference. It may be helpful in preparing informal checklists or writing articles on birds of the region. The taxonomic sequence & the scientific names in the SORL largely follow the International Ornithological Congress (IOC) List at www.worldbirdnames.org. We have departed from this source when new research has revealed new understanding or when we have decided that other English names are more appropriate for the OSME Region. The English names in the SORL include many informal names as denoted thus '…' in the ORL. The SORL uses subspecific names where useful; eg where diagnosable populations appear to be approaching species status or are species whose subspecies might be elevated to full species (indicated by round brackets in scientific names); for now, we remain neutral on the precise status - species or subspecies - of such taxa. Future research may amend or contradict our presentation of the SORL; such changes will be incorporated in succeeding SORL versions. This checklist was devised and prepared by AbdulRahman al Sirhan, Steve Preddy and Mike Blair on behalf of OSME Council. Please address any queries to [email protected]. -

Pico Mediano – Dendropicos Medius (Linnaeus, 1758)
Domínguez, J., Ciudad, C. (2017). Pico mediano – Dendropicos medius. En: Enciclopedia Virtual de los Vertebrados Españoles. Salvador, A., Morales, M. B. (Eds.). Museo Nacional de Ciencias Naturales, Madrid. http://www.vertebradosibericos.org/ Pico mediano – Dendropicos medius (Linnaeus, 1758) Jon Domínguez Lacertida. Biodiversidad e Impacto Ambiental Calle Cid 3. CP 02002, Albacete Carlos Ciudad ETSI Montes, Forestal y del Medio Natural Universidad Politécnica de Madrid Ciudad Universitaria s/n 28040 Madrid Versión 29-06-2017 Versiones anteriores: 31-08-2010; 28-09-2016 © J. García Pérez ENCICLOPEDIA VIRTUAL DE LOS VERTEBRADOS ESPAÑOLES Sociedad de Amigos del MNCN – MNCN - CSIC Domínguez, J., Ciudad, C. (2017). Pico mediano – Dendropicos medius. En: Enciclopedia Virtual de los Vertebrados Españoles. Salvador, A., Morales, M. B. (Eds.). Museo Nacional de Ciencias Naturales, Madrid. http://www.vertebradosibericos.org/ Nombres vernáculos Castellano: pico mediano, Catalán: picot garser mitjà, Euskera: okil ertaina, Gallego: peto mediano (Clavell et al., 2005). Alemán: Mittelspecht, Francés: Pic mar, Inglés: Middle Spotted Woodpecker, Italiano: Picchio rosso mezzano, Portugués: Pica-pau-mediano (Lepage, 2009). Filogenia Incluido tradicionalmente en el género Dendrocopos, un estudio de ADN mitocondrial basado en citocromo b y 12SrRNA asignó al género Leiopicus las especies medius, auriceps y mahrattensis (Winkler et al., 2014). Otro estudio, basado en intrones nucleares y un gen mitocondrial, ha corroborado que el género Dendrocopos no es monofilético y ha propuesto restringir el género Leiopicus a la especie mahrattensis e incluir en el género Dendrocoptes a las especies auriceps, medius y dorae (Fuchs y Pons, 2015). Sin embargo, el reconocimiento de Leiopicus mahrattensis hace que Dendropicos en su análisis sea parafilético y poco soportado. -

Birds and Tigers of Northern India
Dusky Eagle Owl on a nest at Keoladeo Ghana N.P. (all photos by Dave Farrow unless otherwise indicated) BIRDS AND TIGERS OF NORTHERN INDIA 21 NOVEMBER – 8 DECEMBER 2016 LEADER: DAVE FARROW This year’s ‘Birds and Tigers of Northern India’ tour was once again a very successful visual feast of avian delights. This tour is full of regional specialities and Indian subcontinent endemics, and among the many highlights were a total of 53 individual Owls seen of 9 species, including Dusky Eagle Owl on a nest, four Tawny Fish Owls and four Brown Fish Owls. We had great fortune with gamebirds, with three Cheer Pheasants plus stunning views of a pair of Koklass Pheasant, plus many Kalij Pheasants, Painted Spurfowl 1 BirdQuest Tour Report: Birds and Tigers of Northern India www.birdquest-tours.com and Jungle Bush-Quail. We also saw Ibisbill, Red-naped Ibis, Black-necked Stork, Sarus Cranes, Indian, Himalayan and Red-headed Vulture, Pallas's and Lesser Fish Eagles, Brown Crake, Indian and Great Stone- curlew, Yellow-wattled and White-tailed Lapwing, Black-bellied and River Tern, Painted and Chestnut-bellied Sandgrouse, and 15 species of Woodpecker including Great Slaty, Himalayan Pied, White-naped and Himalayan Flameback. We found plenty of Slaty-headed and Plum-headed Parakeet, Black-headed Jay, a Rufous-tailed Lark, Indian Bush Lark, the holy trinity of Nepal, Pygmy and Scaly-bellied Wren-Babblers, plus Brook’s Leaf Warbler, Black-faced and Booted Warbler, Black-chinned Babbler, six species of Laughingthrush including Rufous-chinned, Chestnut-bellied and White-tailed Nuthatch, Wallcreeper, Chestnut and Black-throated Thrushes, White-tailed Rubythroat, Golden Bush Robin, dapper Spotted Forktails, Blue-capped Redstart, Variable Wheatear, Fire-tailed Sunbird, Black-breasted Weaver, Altai Accentor, Brown Bullfinch, Blyth’s Rosefinch (a write-in), Crested, White-capped and Red-headed Bunting. -

Northern India: Tigers, Birds and the Himalayas Trip Report October 2017
NORTHERN INDIA: TIGERS, BIRDS AND THE HIMALAYAS TRIP REPORT OCTOBER 2017 By Andy Walker A gorgeous Indian endemic, the Painted Spurfowl, was seen well during the tour. www.birdingecotours.com [email protected] 2 | T R I P R E P O R T India: Tigers, Birds and the Himalayas 2017 This was a customized version of our usual northern India tour, scheduled for January. This tour for Charley and Paul commenced on the 16th of October 2017 at Ranthambhore and concluded in New Delhi on the 30th October 2017. Prior to the tour Charley and Paul took a pre-tour exploring New Delhi and Jaipur and had a one-day extension in New Delhi for some birding there. The tour visited the world-famous Ranthambhore, Keoladeo Ghana (formerly known as Bharatpur Bird Sanctuary), Corbett, and Sultanpur National Parks and spent time in the breathtaking scenery of the Himalayan foothills at Pangot and Sattal. A visit to this part of India would not be complete without taking in the majestic UNESCO World Heritage Sites of Fatehpur Sikri and the Taj Mahal, and so we visited these also. India is well known for its amazing food, and we sampled a great deal of interesting and tasty local dishes throughout the tour. The above combined makes for a perfect Indian birding tour. The tour connected with many exciting birds, such as Indian Skimmer, Indian Courser, Kalij, Koklass, and Cheer Pheasants, Painted Spurfowl, Indian Spotted Eagle, Bearded (Lammergeier), Red-headed, Indian, and Himalayan Vultures, Collared Falconet, Sarus Crane, Black-necked Stork, Small Pratincole, Painted Sandgrouse, Brown Fish Owl, Oriental Scops Owl, Black-bellied and River Terns, Blue-bearded Bee-eater, Great Hornbill, Spotted Forktail, Grey-winged Blackbird, Long-billed and Scaly Thrushes, Himalayan and Siberian Rubythroats, Ultramarine Flycatcher, Striated and Rufous- chinned Laughingthrushes, Grey-crowned Prinia, White-browed Bush Chat, and over 1,600 Red-headed Buntings. -

Changes in Species Composition of Birds and Declining Number of Breeding Territories Over 40 Years in a Nature Conservation Area in Southwest Germany
diversity Article Changes in Species Composition of Birds and Declining Number of Breeding Territories over 40 Years in a Nature Conservation Area in Southwest Germany Fabian Etienne Schrauth and Michael Wink * ID Institute of Pharmacy and Molecular Biotechnology, Heidelberg University, INF 364, D-69120 Heidelberg, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-6221-544880 Received: 6 June 2018; Accepted: 28 August 2018; Published: 30 August 2018 Abstract: Global loss of biodiversity is occurring at an alarming rate and is a major issue in current times. Long-term studies offer the possibility to analyse changes in biodiversity and allow assessments of anthropogenic interventions in ecosystems. At present, various studies in most countries show partially strong declines of insect populations. Due to their role as a food source for many organisms it is assumed that declines of insect abundance might have effects on higher trophic levels like insectivorous birds. For reliable statements on relationships between food availability and population trends, systematic and extensive records of breeding birds are necessary. In this study, we analysed the changes in the range of species, biodiversity, and abundance of a breeding bird community over 43 years in a large nature conservation area in southwest Germany (“Lampertheimer Altrhein” near Mannheim). Since 1974, considerable changes in the spectrum of breeding birds have been found, but the overall biodiversity index did not change. Furthermore, 70% of the investigated species showed decreasing numbers of breeding bird territories, and the overall number of territories across species declined by more than 65%. A classification based on the main diet during the breeding period and habitat use revealed strong declines for insectivorous birds in the study area, especially in wetland and open cultivated landscapes. -

Middle Spotted Woodpecker
Ecological considerations to conciliate forest activities and conservation of the MIDDLE SPOTTED WOODPECKER This document has been written as part of the action 4.1 included in the project POCTEFA Habios EFA 079/15 “Preserving and managing the habitats for avian bioindicators in the Pyrenees”. This project has been 65% co-financed by the European Regional Development Fund (ERDF) through the Spain-France-Andorra V-A Interreg Programme (POCTEFA 2014- 2020). The POCTEFA programme intends to strengthen the social and economic integration of the Spain-France-Andorra cross-border area. It aims to provide financial support for the implementation of cross-border economic, social and environmental projects by promoting joint strategies that foster sustainable territorial development. Partners Associate beneficiaries Co-funding European Regional Development Fund (ERDF) Authors: Hugo Robles, Carlos Ciudad and José María Fernández-García. Photos: Mikel Arrazola/Irekia (back cover, CC license), Carlos Ciudad, José María Fernández- García, Hazi Foundation, iStock.com/LuCaAr, Hugo Robles, Gianluca Roncalli, Jonathan Rubines and Frank Vassen (front cover, CC license). Edition: Hazi Foundation. Design and outline: © Centre de Ciència i Tecnologia Forestal de Catalunya. English review: Brian Webster Recommended citation: Robles, H., Ciudad, C. & Fernández-García, J. M. 2021. Ecological considerations to conciliate forest activities and conservation of the Middle Spotted Woodpecker. POCTEFA Habios project. The opinions in this document belong to their authors and do not necessarily reflect the point of view of the European Commission or any other institution involved in the Habios project. Acknowledgments Much of the knowledge shared in this document comes from long-term studies carried out in the Cantabrian (León and Palencia provinces) and the Basque mountains (Álava province), in northern Spain. -

2020 Indian & Western Himalaya Bird Tour Species List
Eagle-Eye Tours 2020 Western India and Himalaya Guide: Rudolf Koes BIRD SPECIES Seen/ Common Name Scientific Name Heard ANSERIFORMES: Anatidae 1 Lesser Whistling-Duck Dendrocygna javanica s 2 Bar-headed Goose Anser indicus s 3 Graylag Goose Anser anser s 3a Domestic goose Anser sp. s 4 Knob-billed Duck Sarkidiornis melanotos s 5 Ruddy Shelduck Tadorna ferruginea s 6 Cotton Pygmy-Goose Nettapus coromandelianus s 7 Garganey Spatula querquedula s 8 Northern Shoveler Spatula clypeata s 9 Gadwall Mareca strepera s 10 Eurasian Wigeon Mareca penelope s 11 Indian Spot-billed Duck Anas poecilorhyncha s 12 Northern Pintail Anas acuta s 13 Green-winged Teal Anas crecca s 14 Red-crested Pochard Netta rufina s 15 Common Pochard Aythya ferina s 16 Ferruginous Duck Aythya nyroca s 17 Tufted Duck Aythya fuligula s GALLIFORMES: Phasianidae 18 Indian Peafowl Pavo cristatus s 19 Hill Partridge Arborophila torqueola h 20 Common Quail Coturnix coturnix s 21 Black Francolin Francolinus francolinus s 22 Gray Francolin Francolinus pondicerianus s 23 Red Junglefowl Gallus gallus s 24 Kalij Pheasant Lophura leucomelanos s 25 Koklass Pheasant Pucrasia macrolopha h PHOENICOPTERIFORMES: Phoenicopteridae 26 Greater Flamingo Phoenicopterus roseus s PODICIPEDIFORMES: Podicipedidae 27 Little Grebe Tachybaptus ruficollis s 28 Eared Grebe Podiceps nigricollis s COLUMBIFORMES: Columbidae 29 Rock Pigeon Columba livia s 30 Oriental Turtle-Dove Streptopelia orientalis s 31 Eurasian Collared-Dove Streptopelia decaocto s 32 Red Collared-Dove Streptopelia tranquebarica s 33 Spotted -

Albania the Land of Biodiversity
ALBANIA the land of biodiveristy photo Mirian Topi Sustainable nature based tourism for the wellbeing of nature and people 1 Albanian Trip Introductory Nature Tourism in Albania Birding Edition Albanian Trip www.albaniantrip.com [email protected] albanian_trip albanian trip 2 ALBANIA’S NATURAL HERITAGE Albania is situated in south-eastern Europe alongside the Adriatic Sea, sharing borders with Italy in the west, Montenegro in the north-west; Kosovo in the north-east; North Macedonia in the east and Greece in the south. It has a population of around 3 million inhabitants. Albania represents a very rich country in terms of biodiversity, considering its small surface. This is due to diverse relief, natural landscapes and habitats. It is reported that Albania counts a total number of 7233 plant taxon, representing 32% of the floristic diversity of Europe. In addition, Albania holds as well, very rich fauna diversity with a confirmed number of over 5450 species of different groups, among which around 110 species are rare and with high conservation interest at global level. For conservation of country’s biodiversity and natural heritage, Albania has established its National Network of Protected Areas, which includes a number of 800 protected areas (including here the monuments of nature), occupying around 16% of the total territory of the country. 3 One part of the National Network of Protected Areas and also considerable number of other high nature value ecosystems, are part of different international ecological networks. Prespa Lakes, Karavasta Lagoon, Butrinti Lake – Çuka Channel – Stillo Cape and Skader Lake – Buna River are the four Albanian wetlands of international importance recognized by the RAMSAR Convention, ratified by Albania in year 1996.