Genome Size Evolution and Dynamics in Iris, with Special Focus on the Section Oncocyclus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
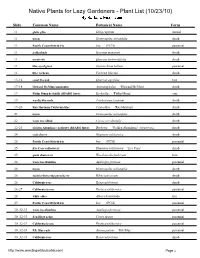
Native Plants for Lazy Gardeners - Plant List (10/23/10)
Native Plants for Lazy Gardeners - Plant List (10/23/10) Slide Common Name Botanical Name Form 11 globe gilia Gilia capitata annual 11 toyon Heteromeles arbutifolia shrub 11 Pacific Coast Hybrid iris Iris (PCH) perennial 11 goldenbush Isocoma menziesii shrub 11 scrub oak Quercus berberidifolia shrub 11 blue-eyed grass Sisyrinchium bellum perennial 11 lilac verbena Verbena lilacina shrub 13-16 coast live oak Quercus agrifolia tree 17-18 Howard McMinn man anita Arctostaphylos 'Howard McMinn' shrub 19 Philip Mun keckiella (RSABG Intro) Keckiella 'Philip Munz' ine 19 woolly bluecurls Trichostema lanatum shrub 19-20 Ray Hartman California lilac Ceanothus 'Ray Hartman' shrub 21 toyon Heteromeles arbutifolia shrub 22 western redbud Cercis occidentalis shrub 22-23 Golden Abundance barberry (RSABG Intro) Berberis 'Golden Abundance' (MAHONIA) shrub 2, coffeeberry Rhamnus californica shrub 25 Pacific Coast Hybrid iris Iris (PCH) perennial 25 Eve Case coffeeberry Rhamnus californica '. e Case' shrub 25 giant chain fern Woodwardia fimbriata fern 26 western columbine Aquilegia formosa perennial 26 toyon Heteromeles arbutifolia shrub 26 fuchsia-flowering gooseberry Ribes speciosum shrub 26 California rose Rosa californica shrub 26-27 California fescue Festuca californica perennial 28 white alder Alnus rhombifolia tree 29 Pacific Coast Hybrid iris Iris (PCH) perennial 30 032-33 western columbine Aquilegia formosa perennial 30 032-33 San Diego sedge Carex spissa perennial 30 032-33 California fescue Festuca californica perennial 30 032-33 Elk Blue rush Juncus patens '.l1 2lue' perennial 30 032-33 California rose Rosa californica shrub http://www weedingwildsuburbia com/ Page 1 30 032-3, toyon Heteromeles arbutifolia shrub 30 032-3, fuchsia-flowering gooseberry Ribes speciosum shrub 30 032-3, Claremont pink-flowering currant (RSA Intro) Ribes sanguineum ar. -

Ecological Response of Some Iris L. Taxa (Iridaceae) in Turkey
Bangladesh J. Bot. 40(2): 177-184, 2011 (December) ECOLOGICAL RESPONSE OF SOME IRIS L. TAXA (IRIDACEAE) IN TURKEY 1 2 NEZAHAT KANDEMIR*, ALI ÇELIK AND ABDULKADIR SÜRÜCÜ Department of Biology, Education Faculty, Amasya University, Amasya-Turkey Key words: Iris taxa, Endemic, Rare, Ecological properties, Relationship Abstract Ecological relationships of some Iris taxa belonging to subgenera Hermodactyloides (I. danfordiae, I. histrio subsp. aintabensis, I. histrio subsp. histrio, I. reticulata, I. bakeriana, I. pamphylica) and Scorpiris (I. aucheri and I. persica) have been compared and relationships between taxa have been determined. These taxa are geophytes and flower in Spring. I. danfordiae, I. pamphylica and subsp. aintabensis are endemic to Turkey. I. reticulata and I. persica have widespread distribution while I. aucheri, subsp. aintabensis, subsp. histrio, I. pamphylica and I. bakeriana have restricted distribution in Turkey. Soil samples of the taxa were collected during flowering periods and physical and chemical properties (texture class, total salinity %, pH, CaCO3 %, organic matter %, N %, P kg/da, K kg/da, Ca , Mg, Mn, Cu, Fe and Zn ppm) were determined. The correlations between the soil analyses and taxa were evaluated using regression analysis. The P and CaCO3 values were found to be more effective than the other soil factors in the distributions of investigated taxa. Introduction Iris L. is one of the largest genus of Iridaceae family and comprises over 300 species in the world. They have been distributed in the Northern Hemisphere (Yu et al. 2009). Iris species have been used as ornamental plants in vegetative landscape of the parks and gardens in many countries since ancient times because of very beautiful and colorful flowers (specially in rock garden I. -

Philip J. Savage, Jr
ISSUE 73 M4SNOU4 Philip J. Savage, Jr. May 8, 1917 — October 13, 2002 Phil Savnge, /r. , an icon m the Magnolia worhl, passed au ay last October of conr plica- lions from the West Nile virus. Phil, a renotvrred magnolin Irybcidrzer, specialized in breeding mngnolias that u&cre cold Irardy. Becnuse of this, nragnolia entirusiasts living in colder climalcs now have morry rrrorc choices thnn did Dennis Ledvh rn u&hen he bought Iris lrouse irr Green Bay, Wiscorrsin in thc Intr typos Plril hns left an errduring legaclt n&itlr tire mnny /Inc, cold-lurrdy hybrids Ire bred. Following arc set&eral rwni niscclrces fronr irrdividuals who toere deqviy in/luencerl by Phil mrd Iris u&ork u&ith mngrrolins. DENNIs LEDVINA WIUTES. Back in the late yos the landscaping around my new house consisted of three magnolias: two M. x sorrlarrgeana, and a M. x loebneri 'MerrilL' At the time, these were the only magnolias gener- ally available at local nurseries. As I watched these magnolias bloom each spring, I became more intrigued with their beautiful Bowers and began driv- ing around Green Bay to observe and admire some of the established trees. My admiration for the genus contin- ued to grow each year as I began col- lecting more information about these magnificent plants. One summer I was in the Detroit area and I decided to call this magnolia expert, Phil Savage, that I had read so much about. I can vividly remember calling Phil from a telephone booth on Telegraph Road and finding mysell, an unknown amateur, talking to a magnolia expert who from the first made me feel like a lifelong friend. -

1 Acanthus Dioscoridis Acanthaceae 2 Blepharis Persica Acanthaceae 3
Row Species Name Family 1 Acanthus dioscoridis Acanthaceae 2 Blepharis persica Acanthaceae 3 Acer mazandaranicum Aceraceae 4 Acer monspessulanum subsp. persicum Aceraceae 5 Acer monspessulanum subsp. assyriacum Aceraceae 6 Acer monspessulanum subsp. cinerascens Aceraceae 7 Acer monspessulanum subsp. turcomanicum Aceraceae 8 Acer tataricum Aceraceae 9 Acer campestre Aceraceae 10 Acer cappadocicum Aceraceae 11 Acer monspessulanum subsp. ibericum Aceraceae 12 Acer hyrcanum Aceraceae 13 Acer platanoides Aceraceae 14 Acer velutinum Aceraceae 15 Aizoon hispanicum Aizoaceae 16 Mesembryanthemum nodiflorum Aizoaceae 17 Sesuvium verrucosum Aizoaceae 18 Zaleya govindia Aizoaceae 19 Aizoon canariense Aizoaceae 20 Alisma gramineum Alismataceae 21 Damasonium alisma Alismataceae 22 Alisma lanceolatum Alismataceae 23 Alisma plantago-aquatica Alismataceae 24 Sagittaria trifolia Alismataceae 25 Allium assadii Alliaceae Row Species Name Family 26 Allium breviscapum Alliaceae 27 Allium bungei Alliaceae 28 Allium chloroneurum Alliaceae 29 Allium ellisii Alliaceae 30 Allium esfandiarii Alliaceae 31 Allium fedtschenkoi Alliaceae 32 Allium hirtifolium Alliaceae 33 Allium kirindicum Alliaceae 34 Allium kotschyi Alliaceae 35 Allium lalesaricum Alliaceae 36 Allium longivaginatum Alliaceae 37 Allium minutiflorum Alliaceae 38 Allium shelkovnikovii Alliaceae 39 Allium subnotabile Alliaceae 40 Allium subvineale Alliaceae 41 Allium wendelboi Alliaceae 42 Nectaroscordum koelzii Alliaceae 43 Allium akaka Alliaceae 44 Allium altissimum Alliaceae 45 Allium ampeloprasum subsp. -

May 15, 2016 Passing Peony and Iris Plants on from Generation to Generation Annette Meyer Heisdorffer Daviess County Extension Agent for Horticulture
May 15, 2016 Passing Peony and Iris Plants on from Generation to Generation Annette Meyer Heisdorffer Daviess County Extension Agent for Horticulture After lunch on Mother’s Day, my mom and I surveyed her garden, especially the peonies. We both agreed that I needed to propagate her peonies and plant them in my garden. These are special, because I remember them growing in my grandmother’s garden. Peonies are commonly passed down from generation to generation. My goal is to someday share them with my twins. Our discussion included the irises, which are another heritage plant. Both plants are blooming beautifully in May and are spectacular in the garden. Information about these two plants will be provided in this article. Peony (Paeonia officinalis, Paeonia lactiflora, and hybrids) is a herbaceous perennial, which means at the end of the growing season it will die back to the ground. However, the plant returns year after year. Peonies grow best in full sun and well-drained soil. There are tree peonies (Paeonia suffruticosa) which have a woody stem, but those are not as common and require different growing conditions. The tree peony will not be discussed here. According to Dr. Rick Durham, Extension Specialist for Consumer Horticulture, peonies can be found in landscapes across Kentucky. Peonies have a long life span and are commonly grown in the garden. When planting the root, make sure it is not too deep. The eyes or bud should be just below the surface of the soil. If it is planted too deeply, the plants won't bloom. -

Basic Guide to the Culture of Peonies This Is Easily Done by Pulling Them Downwards and Sideways with the Fingers
A handful of sheep manure to a plant may be given in the spring to improve the bloom. Liquid manure also may be used with discretion, for the same purpose. Disbudding. Most varieties of peonies develop several small lateral or America’s Largest Grower of Daylilies, Peonies and Iris sidebuds near the base of the terminal bud. If large flowers are wanted, the side buds should be removed so the strength will all go into the terminal bud. The side-buds should be removed as soon as they are about the size of a pea. Basic Guide to the Culture of Peonies This is easily done by pulling them downwards and sideways with the fingers. o Some people prefer leaving their side-buds which develop and prolong the P.O. Box 338 • Sarcoxie, MO 64862 • (417) 548-3514 blooming season. The side-buds bloom later than the terminal buds. Insect Pests. In some sections of the country, where thrips are Basic Guide to the Culture of Peonies prevalent, some late varieties are damaged to the extent that the buds fail to open even after they are almost fully developed. Spraying or dusting, at Peonies are easily grown and their requirements are few, but they respond weekly intervals should control the thrips very well. Apply first application beautifully to a little special care and attention by producing best quality when buds are about the size of a large marble. We like Orthene. flowers and many of them. With this thought in mind we offer the following suggestions gained from many years experience in growing and Planted too deeply...examine and if Why Peonies Do Not Bloom. -

STEVEN and ERIN FORD GARDEN Botanical Name Common Name
STEVEN AND ERIN FORD GARDEN Botanical Name Common Name Abutilon Flowering Maple Acer buergerianum Trident Maple Acer japonicum Full Moon Maple Acer palmatum Japanese Maple Acer palmatum 'Bloodgood' Japanese Maple Achillea millefolium Yarrow Adiantum Maidenhair Fern Agapanthus Lily of the Nile Ajuga Alcea rosea Ajuga Alcea rosea Hollyhock Anemone japonica Japanese Anemone Antirrhinum majus Snapdragon Aquilegia Columbine Aralia Japanese Aralia Artemisia stelleriana Dusty Miller Artemisia stelleriana Powis Castle Artemisia Asparagus densiflorus Asparagus Fern Asparagus densiflorus 'Sprengeri' Sprenger Asparagus Fern Asparagus retrofractus Ming Fern Azalea Azalea Azalea 'Hino' Basil Begonia Berberis thumbergii 'Golden Nugget' Japanese Barberry Bletilla striata Chinese Ground Orchid Boysenberries Buddleja Butterfly Bush Buxus sempervirens Common Boxwood Calendula officinalis Pot Marigold Camellia japonica Camellia sasanqua Carex oshimensis Sedge Carex variegata Sedge Caryopteris Bluebird Ceanothus Cedrus deodara Deodar Cedar Centranthus ruber Jupiter's Beard Cercis canadensis Eastern Rudbud Chives Chrysanthemum Chrysanthemum maximum Shasta Daisy Citrus Dwarf Blood Orange Citrus Dwarf Grapefruit 1 Citrus Dwarf Tangerine Citrus Navel Orange Citrus Variegated Lemon Clematis Cornus Dogwood Cornus kousa Kousa Dogwood Cornus stolonifera Redtwig Dogwood Cotinus Smoke Tree Cryptomeria japonica Japanese Cryptomeria Cyathea cooperi Australian Tree Fern Cyclamen Delphinium Dianella tasmanica 'Yellow Stripe' Flax Lily Dianthus Pink Dianthus barbatus Sweet -

These De Doctorat De L'universite Paris-Saclay
NNT : 2016SACLS250 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY, préparée à l’Université Paris-Sud ÉCOLE DOCTORALE N° 567 Sciences du Végétal : du Gène à l’Ecosystème Spécialité de doctorat (Biologie) Par Mlle Nour Abdel Samad Titre de la thèse (CARACTERISATION GENETIQUE DU GENRE IRIS EVOLUANT DANS LA MEDITERRANEE ORIENTALE) Thèse présentée et soutenue à « Beyrouth », le « 21/09/2016 » : Composition du Jury : M., Tohmé, Georges CNRS (Liban) Président Mme, Garnatje, Teresa Institut Botànic de Barcelona (Espagne) Rapporteur M., Bacchetta, Gianluigi Università degli Studi di Cagliari (Italie) Rapporteur Mme, Nadot, Sophie Université Paris-Sud (France) Examinateur Mlle, El Chamy, Laure Université Saint-Joseph (Liban) Examinateur Mme, Siljak-Yakovlev, Sonja Université Paris-Sud (France) Directeur de thèse Mme, Bou Dagher-Kharrat, Magda Université Saint-Joseph (Liban) Co-directeur de thèse UNIVERSITE SAINT-JOSEPH FACULTE DES SCIENCES THESE DE DOCTORAT DISCIPLINE : Sciences de la vie SPÉCIALITÉ : Biologie de la conservation Sujet de la thèse : Caractérisation génétique du genre Iris évoluant dans la Méditerranée Orientale. Présentée par : Nour ABDEL SAMAD Pour obtenir le grade de DOCTEUR ÈS SCIENCES Soutenue le 21/09/2016 Devant le jury composé de : Dr. Georges TOHME Président Dr. Teresa GARNATJE Rapporteur Dr. Gianluigi BACCHETTA Rapporteur Dr. Sophie NADOT Examinateur Dr. Laure EL CHAMY Examinateur Dr. Sonja SILJAK-YAKOVLEV Directeur de thèse Dr. Magda BOU DAGHER KHARRAT Directeur de thèse Titre : Caractérisation Génétique du Genre Iris évoluant dans la Méditerranée Orientale. Mots clés : Iris, Oncocyclus, région Est-Méditerranéenne, relations phylogénétiques, status taxonomique. Résumé : Le genre Iris appartient à la famille des L’approche scientifique est basée sur de nombreux Iridacées, il comprend plus de 280 espèces distribuées outils moléculaires et génétiques tels que : l’analyse de à travers l’hémisphère Nord. -

Hybrid Fitness, Adaptation and Evolutionary Diversification: Lessons
Heredity (2012) 108, 159–166 & 2012 Macmillan Publishers Limited All rights reserved 0018-067X/12 www.nature.com/hdy REVIEW Hybrid fitness, adaptation and evolutionary diversification: lessons learned from Louisiana Irises ML Arnold, ES Ballerini and AN Brothers Estimates of hybrid fitness have been used as either a platform for testing the potential role of natural hybridization in the evolution of species and species complexes or, alternatively, as a rationale for dismissing hybridization events as being of any evolutionary significance. From the time of Darwin’s publication of The Origin, through the neo-Darwinian synthesis, to the present day, the observation of variability in hybrid fitness has remained a challenge for some models of speciation. Yet, Darwin and others have reported the elevated fitness of hybrid genotypes under certain environmental conditions. In modern scientific terminology, this observation reflects the fact that hybrid genotypes can demonstrate genotypeÂenvironment interactions. In the current review, we illustrate the development of one plant species complex, namely the Louisiana Irises, into a ‘model system’ for investigating hybrid fitness and the role of genetic exchange in adaptive evolution and diversification. In particular, we will argue that a multitude of approaches, involving both experimental and natural environments, and incorporating both manipulative analyses and surveys of natural populations, are necessary to adequately test for the evolutionary significance of introgressive hybridization. An appreciation of the variability of hybrid fitness leads to the conclusion that certain genetic signatures reflect adaptive evolution. Furthermore, tests of the frequency of allopatric versus sympatric/parapatric divergence (that is, divergence with ongoing gene flow) support hybrid genotypes as a mechanism of evolutionary diversification in numerous species complexes. -

Ekonomik Önemi.Pdf
INTRODUCTION OF ECONOMICALLY IMPORTANT BULBOUS PLANTS COLLECTED FROM WILD FLORA IN SEMI ARID CLIMATIC CONDITIONS OF SOUTHEASTERN ANATOLIAN REGION OF TURKEY Süleyman KIZIL1, Khalid Mahmood KHAWAR2 and Neşet ARSLAN2 1 Department of Field Crops, Agriculture Faculty, Dicle University, 21280 Diyarbakir, Turkey 2 Department of Field Crops, Agriculture Faculty, Ankara University, 06100 Ankara, Turkey Corresponding author: [email protected] Abstract Turkey has rich biodiversity due to its topography comprising of plains, plateaus and mountainous regions that have contributed to enrichment of its flora including bulbous plants. Many among these have potential for use in pharmaceutical and ornamental plant industries. However, owing to lack of proper research many among these plants are yet to be evaluated for commercial propagation. Leaves, bulbs and flowers among many plant parts are being evaluated locally as salads, vegetables, products of pharmaceutical importance and flowers for use in cut flower and ornamental plant industries. The study aimed to find economically important plant geophytes that grow in the wild of the South Eastern Anatolian climatic zones. To meet the objective, a field survey of bulbous geophytes of South Eastern and Eastern Anatolia was carried out during April-July periods of 2011 and 2012 years. The survey results indicated distribution of bulb geophytes at altitudes of 640 to 2651m. The geophytes belonging to the genus Allium, Biarum, Bellevalia, Crocus, Eranthis, Fritillaria, Gladiolus, Hyacinthus, Iris, Ixillirion, Muscari, Narcissus, Ornithogalum, Sternbergia, Scilla, Tulipa, Ophyrs and Orchis were collected. After initial screening, it was decided to culture 40 species; the bulbs of these species were planted in the collection gardens of the Department of Field Crops, Dicle University, Diyarbakir for determination of several parameters including, flowering date, duration of flowering time and other agronomical characteristics important for bulbous species. -

Turkish Silk Road Trip Report 2019
TURKISH SILK ROAD TRIP REPORT 2019 1 Day 1 6 May To Goreme We all arrived from various places to Cappadocia. Day 2 7 May Cappadocia I A fine clear morning revealed the remarkable convoluted landscape of Cappadocia – a blend of towers and smooth-eroded hills, some pink some cream. We met with our guide Gaye and set off for a quieter part of this popular region. Our first stop was near a small church and above this the path led to a fine lookout across the landscape including some amazing chimneys capped with dark hats of denser rock. Indeed, it is the rapid erosion of the various layers of compacted ash that have created this landscape, a legacy of the regions intensely volcanic past. There many Alpine Swifts sweeping overhead and a few interesting flowers with tufts of bluish Trigonella coerulescens, Silene conoidea, Euphorbia sp and big patches of Eruca sativa that were a magnet for the many Painted Ladies on the wing. We moved on to another site with an old monastery that still retained some very old frescoes and painted ceilings as well as a very old Seljuk mosque. Here there was plentiful Hypercoum pseudograndiflorum along the paths. Uta exchanged tips on bean cultivation with a local farmer who spoke a smattering of German before we left. Lunch was in a cherry orchard, thronging with butterflies as well as by chance, being next to a nesting Long-eared Owl which peered down at us the whole time we were there. Then it was onto see a special plant, crossing the undulating steppes and wheat fields to an innocuous-looking hill. -

Spatial Distribution of Flower Colour Polymorphism in Iris Lutescens Eric Imbert
Spatial distribution of flower colour polymorphism in Iris lutescens Eric Imbert To cite this version: Eric Imbert. Spatial distribution of flower colour polymorphism in Iris lutescens. Botany Letters, Taylor & Francis, In press, 10.1080/23818107.2020.1833750. hal-02958925 HAL Id: hal-02958925 https://hal.archives-ouvertes.fr/hal-02958925 Submitted on 6 Oct 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Title : Geographical distribution of flower colour polymorphism in Iris lutescens Author: Eric Imbert Address : ISEM, University of Montpellier – Montpellier, France [email protected] Accepté pour publication Botany Letters https://doi.org/10.1080/23818107.2020.1833750 Abstract Iris lutescens is a common species occurring mainly in dry limestone habitats in Western Italy, Southern France and Spain. The species shows a remarkable polymorphism for flower colour, and yellow and purple flowers can be found in the same population. As the species is a deceptive one, the previous studies on the maintenance of such a polymorphism were linked to its pollination ecology. Here, I reported on the spatial distribution of the polymorphism, and showed that Spanish populations are mostly purple monomorphic. In contrast, populations in the South of France and Italy show the complete range, from 0 to 1, for the frequency of yellow morph, and the spatial autocorrelation for morph frequencies is very low.