Facing the Future of Craniofacial Genetics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Turnedhead Adductedhip Truncal Curvature Syndrome
Archives ofDisease in Childhood 1994; 70: 515-519 515 Turned head adducted hip truncal curvature syndrome Arch Dis Child: first published as 10.1136/adc.70.6.515 on 1 June 1994. Downloaded from Chiaki Hamanishi, Seisuke Tanaka Abstract curvature. An epidemiological analysis was One hundred and eight neonates and carried out to determine whether the intra- infants who showed the clinical triad of a uterine environment of these asymmetrically head turned to one side, adduction con- deformed babies had been restricted. The tracture of the hip joint on the occipital clinical course of each feature of the clinical side of the turned head, and truncal cur- triad was also analysed to determine whether vature, which we named TAC syndrome, TAC syndrome is aetiologically related to any were studied. These cases included seven subsequent paediatric disorders. with congenital and five with late infantile dislocations of the hip joint and 14 who developed muscular torticollis. Forty one Patients and methods were among 7103 neonates examined by We studied a total of 108 cases with TAC one of the authors. An epidemiological syndrome. Of them, 41 were among a total analysis confirmed the aetiology of the number of 7103 neonates personally examined syndrome to be environmental. The side by one of the authors (CH) at newborn exami- to which the head was turned and that of nations conducted at hospitals in four cities the adducted hip contracture showed a since 1981. Thirteen were referred neonates. high correlation with the side of the The remaining 54 were among infants aged maternal spine on which the fetus had from 10 days to 3 months who were referred to been lying. -
Basic Concepts in Basic Concepts in Dysmorphology
Basic Concepts in Dysmorphology Samia Temtamy* & Mona Aglan** *Professor of Human Genetics **Professor of Clinical Genetics Human Genetics & Genome Research Division National Research Centre, Cairo, Egypt OtliOutline y Definition of dysmorphology y Definition of terms routinely used in the description of birth defects y Impact of malformations y The difference between major & minor anomalies y Approach to a dysmorphic individual: y Suspicion & analysis y Systematic physical examination y CfitifdiConfirmation of diagnos is y Intervention y Summary 2 DfiitiDefinition of fd dysmorph hlology y The term “dysmorphology” was first coined by Dr. DidSithUSAiDavid Smith, USA in 1960s. y It implies study of human congenital defects and abnormalities of body structure that originate before birth. y The term “dysmorphic” is used to describe individuals whose physical fffeatures are not usually found in other individuals with the same age or ethnic background. y “Dys” (Greek)=disordered or abnormal and “Morph”=shape 3 Definition of terms routinely used in the d escri pti on of bi rth d ef ect s y A malformation / anomaly: is a primary defect where there i s a bas ic a ltera tion o f s truc ture, usuall y occurring before 10 weeks of gestation. y Examples: cleft palate, anencephaly, agenesis of limb or part of a limb. 4 Cleft lip & palate Absence of digits (ectrodactyly) y Malformation Sequence: A pattern of multiple defects resulting from a single primary malformation. y For example: talipes and hydrocephalus can result from a lumbar neural tube defect. Lumbar myelomeningeocele 5 y Malformation Syndrome: A pattern of features, often with an underlying cause, that arises from several different errors in morphogenesis. -

Tracheal Perforation in a Neonate: a Devastating
Letters to Editor Tracheal perforation in a size of endotracheal tube, cuff overinflation, cough and vigorous movements of head and neck.[2,3] neonate: A devastating Neonatal tracheal injury is ascribed to factors like complication following traumatic traumatic delivery, weak trachea, congenital tracheal endotracheal intubation stenosis, ring agenesis, metal stylets, rigid endotracheal tube, excessive external laryngeal pressure and [1,4,5] Sir, prolonged ventilation. Tracheal injury in neonates following endotracheal In our case, rigorous attempts at intubation along with intubation represents an uncommon complication excessive hyperextension of head and neck due to altered rarely described in literature but carries high anatomy are likely to have contributed to tracheal injury. morbidity and a mortality rate of 70%.[1] We describe Multiple attempts at intubation are known to result in a [4] a case of neonatal tracheal perforation following false tract and are related to anterior tracheal lesions. multiple attempts at endotracheal intubation due to an Incidentally, our case was an undiagnosed Pierre unanticipated difficulty in an emergency situation in Robins Sequence. A small receding mandible, tongue an undiagnosed case of Pierre Robin Sequence. immobility and cleft palate are identified as independent factors for difficult airway with a risk of upper airway A 4-week-old female baby presented to the emergency obstruction, difficult mask holding, difficult intubation, department with respiratory difficulty. In view of severe leak through the cleft resulting in inadequate ventilation respiratory distress and desaturation (SpO2- 60%) a as well as passage of the endotracheal tube into the decision was made to provide ventilatory support to cleft.[6] Successful airway management in such situation the neonate. -

Dysmorphology Dysmorphism
Dysmorphology Carolyn Jones, M.D., PhD Dysmorphism Morphologic developmental abnormalities. This may been seen in many syndromes of genetic or environmental origin. 1 Malformation A recognized dysmorphic feature. A structure not formed correctly. This can either be the cause of genetic factors or environment. 2 Deformation An external force resulting in the inability of a structure to form correctly. Example: club feet in a woman with oligohydramnios, fibroid tumors or multiple gestation. Disruption Birth defect resulting from the destruction of a normally forming structure. This can be caused by vascular occlusion, teratogen, or rupture of amniotic sac (amniotic band syndrome). 3 Common Dysmorphic features Wide spacing between eyes, hypertelorism Narrow spacing between eyes, hypotelorism Palpebral fissure length Epicanthal folds Common Dysmorphic Features Philtrum length Upper lip Shape of nose 4 Syndrome A number of malformations seen together Cause of Syndromes Chromosomal aneuploidy Single Gene abnormalities Teratogen exposure Environmental 5 Chromosomal Aneuploidy Nondisjuction resulting in the addition or loss of an entire chromosome Deletion of a part of a chromosome Microdeletion syndromes (small piece missing which is usually only detected using special techniques) 6 Common Chromosomal Syndromes caused by Nondisjuction Down syndrome (trisomy 21) Patau syndrome (trisomy 13) Edwards syndrome (trisomy 18) Turner syndrome (monosomy X) Klinefelter syndrome (47,XXY) Clinical Features at birth of Down syndrome Low set small ears Hypotonia Simian crease Wide space between first and second toe Flat face 7 Clinical Features of Down Syndrome Small stature Congenital heart defects (50-70%) Acquired and congenital hearing impairment. Duodenal atresia Hirshprungs disease Trisomy 13 Occurs in 1/5000 live births. -
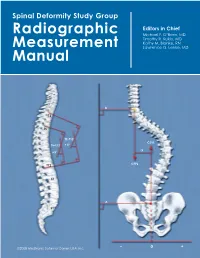
Spinal Deformity Study Group
Spinal Deformity Study Group Editors in Chief Radiographic Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Measurement Lawrence G. Lenke, MD Manual B T2 T5 T2–T12 CSVL T5–T12 +X° -X +X° C7PL T12 L2 A S1 ©2008 Medtronic Sofamor Danek USA, Inc. – 0 + Radiographic Measurement Manual Editors in Chief Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Lawrence G. Lenke, MD Section Editors Keith H. Bridwell, MD Kathy M. Blanke, RN Christopher L. Hamill, MD William C. Horton, MD Timothy R. Kuklo, MD Hubert B. Labelle, MD Lawrence G. Lenke, MD Michael F. O’Brien, MD David W. Polly Jr, MD B. Stephens Richards III, MD Pierre Roussouly, MD James O. Sanders, MD ©2008 Medtronic Sofamor Danek USA, Inc. Acknowledgements Radiographic Measurement Manual The radiographic measurement manual has been developed to present standardized techniques for radiographic measurement. In addition, this manual will serve as a complimentary guide for the Spinal Deformity Study Group’s radiographic measurement software. Special thanks to the following members of the Spinal Deformity Study Group in the development of this manual. Sigurd Berven, MD Hubert B. Labelle, MD Randal Betz, MD Lawrence G. Lenke, MD Fabien D. Bitan, MD Thomas G. Lowe, MD John T. Braun, MD John P. Lubicky, MD Keith H. Bridwell, MD Steven M. Mardjetko, MD Courtney W. Brown, MD Richard E. McCarthy, MD Daniel H. Chopin, MD Andrew A. Merola, MD Edgar G. Dawson, MD Michael Neuwirth, MD Christopher DeWald, MD Peter O. Newton, MD Mohammad Diab, MD Michael F. -

Orthopedic-Conditions-Treated.Pdf
Orthopedic and Orthopedic Surgery Conditions Treated Accessory navicular bone Achondroplasia ACL injury Acromioclavicular (AC) joint Acromioclavicular (AC) joint Adamantinoma arthritis sprain Aneurysmal bone cyst Angiosarcoma Ankle arthritis Apophysitis Arthrogryposis Aseptic necrosis Askin tumor Avascular necrosis Benign bone tumor Biceps tear Biceps tendinitis Blount’s disease Bone cancer Bone metastasis Bowlegged deformity Brachial plexus injury Brittle bone disease Broken ankle/broken foot Broken arm Broken collarbone Broken leg Broken wrist/broken hand Bunions Carpal tunnel syndrome Cavovarus foot deformity Cavus foot Cerebral palsy Cervical myelopathy Cervical radiculopathy Charcot-Marie-Tooth disease Chondrosarcoma Chordoma Chronic regional multifocal osteomyelitis Clubfoot Congenital hand deformities Congenital myasthenic syndromes Congenital pseudoarthrosis Contractures Desmoid tumors Discoid meniscus Dislocated elbow Dislocated shoulder Dislocation Dislocation – hip Dislocation – knee Dupuytren's contracture Early-onset scoliosis Ehlers-Danlos syndrome Elbow fracture Elbow impingement Elbow instability Elbow loose body Eosinophilic granuloma Epiphyseal dysplasia Ewing sarcoma Extra finger/toes Failed total hip replacement Failed total knee replacement Femoral nonunion Fibrosarcoma Fibrous dysplasia Fibular hemimelia Flatfeet Foot deformities Foot injuries Ganglion cyst Genu valgum Genu varum Giant cell tumor Golfer's elbow Gorham’s disease Growth plate arrest Growth plate fractures Hammertoe and mallet toe Heel cord contracture -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -

E Proteins and ID Proteins: Helix-Loop-Helix Partners in Development and Disease
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Developmental Cell Review E Proteins and ID Proteins: Helix-Loop-Helix Partners in Development and Disease Lan-Hsin Wang1 and Nicholas E. Baker1,2,3,* 1Department of Genetics, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA 2Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA 3Department of Ophthalmology and Visual Sciences, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA *Correspondence: [email protected] http://dx.doi.org/10.1016/j.devcel.2015.10.019 The basic Helix-Loop-Helix (bHLH) proteins represent a well-known class of transcriptional regulators. Many bHLH proteins act as heterodimers with members of a class of ubiquitous partners, the E proteins. A widely expressed class of inhibitory heterodimer partners—the Inhibitor of DNA-binding (ID) proteins—also exists. Genetic and molecular analyses in humans and in knockout mice implicate E proteins and ID proteins in a wide variety of diseases, belying the notion that they are non-specific partner proteins. Here, we explore relationships of E proteins and ID proteins to a variety of disease processes and highlight gaps in knowledge of disease mechanisms. E proteins and Inhibitor of DNA-binding (ID) proteins are widely conferring DNA-binding specificity and transcriptional activation expressed transcriptional regulators with very general functions. on heterodimers with the ubiquitous E proteins (Figure 1). They are implicated in diseases by evidence ranging from Another class of pervasive HLH proteins acts in opposition to confirmed Mendelian inheritance, association studies, and E proteins. -

Craniosynostosis and Related Disorders Mark S
CLINICAL REPORT Guidance for the Clinician in Rendering Pediatric Care Identifying the Misshapen Head: Craniosynostosis and Related Disorders Mark S. Dias, MD, FAAP, FAANS,a Thomas Samson, MD, FAAP,b Elias B. Rizk, MD, FAAP, FAANS,a Lance S. Governale, MD, FAAP, FAANS,c Joan T. Richtsmeier, PhD,d SECTION ON NEUROLOGIC SURGERY, SECTION ON PLASTIC AND RECONSTRUCTIVE SURGERY Pediatric care providers, pediatricians, pediatric subspecialty physicians, and abstract other health care providers should be able to recognize children with abnormal head shapes that occur as a result of both synostotic and aSection of Pediatric Neurosurgery, Department of Neurosurgery and deformational processes. The purpose of this clinical report is to review the bDivision of Plastic Surgery, Department of Surgery, College of characteristic head shape changes, as well as secondary craniofacial Medicine and dDepartment of Anthropology, College of the Liberal Arts characteristics, that occur in the setting of the various primary and Huck Institutes of the Life Sciences, Pennsylvania State University, State College, Pennsylvania; and cLillian S. Wells Department of craniosynostoses and deformations. As an introduction, the physiology and Neurosurgery, College of Medicine, University of Florida, Gainesville, genetics of skull growth as well as the pathophysiology underlying Florida craniosynostosis are reviewed. This is followed by a description of each type of Clinical reports from the American Academy of Pediatrics benefit from primary craniosynostosis (metopic, unicoronal, bicoronal, sagittal, lambdoid, expertise and resources of liaisons and internal (AAP) and external reviewers. However, clinical reports from the American Academy of and frontosphenoidal) and their resultant head shape changes, with an Pediatrics may not reflect the views of the liaisons or the emphasis on differentiating conditions that require surgical correction from organizations or government agencies that they represent. -

Spondylolysis and Spondylolisthesis in Children and Adolescents: I
Spondylolysis and Spondylolisthesis in Children and Adolescents: I. Diagnosis, Natural History, and Nonsurgical Management Ralph Cavalier, MD Abstract Martin J. Herman, MD Spondylolysis and spondylolisthesis are often diagnosed in children Emilie V. Cheung, MD presenting with low back pain. Spondylolysis refers to a defect of Peter D. Pizzutillo, MD the vertebral pars interarticularis. Spondylolisthesis is the forward translation of one vertebral segment over the one beneath it. Isth- Dr. Cavalier is Attending Orthopaedic mic spondylolysis, isthmic spondylolisthesis, and stress reactions Surgeon, Summit Sports Medicine and involving the pars interarticularis are the most common forms seen Orthopaedic Surgery, Brunswick, GA. Dr. Herman is Associate Professor, in children. Typical presentation is characterized by a history of Department of Orthopaedic Surgery, activity-related low back pain and the presence of painful spinal Drexel University College of Medicine, mobility and hamstring tightness without radiculopathy. Plain ra- St. Christopher’s Hospital for Children, Philadelphia, PA. Dr. Cheung is Fellow, diography, computed tomography, and single-photon emission Department of Orthopaedic Surgery, computed tomography are useful for establishing the diagnosis. Mayo Clinic, Rochester, MN. Dr. Symptomatic stress reactions of the pars interarticularis or adjacent Pizzutillo is Professor, Department of vertebral structures are best treated with immobilization of the Orthopaedic Surgery, Drexel University College of Medicine, St. Christopher’s spine and activity restriction. Spondylolysis often responds to brief Hospital for Children. periods of activity restriction, immobilization, and physiotherapy. None of the following authors or the Low-grade spondylolisthesis (≤50% translation) is treated similarly. departments with which they are The less common dysplastic spondylolisthesis with intact posterior affiliated has received anything of value elements requires greater caution. -

Inhibition of SNW1 Association with Spliceosomal Proteins Promotes
Cancer Medicine Open Access ORIGINAL RESEARCH Inhibition of SNW1 association with spliceosomal proteins promotes apoptosis in breast cancer cells Naoki Sato1, Masao Maeda2, Mai Sugiyama2, Satoko Ito2, Toshinori Hyodo2, Akio Masuda3, Nobuyuki Tsunoda1, Toshio Kokuryo1, Michinari Hamaguchi2, Masato Nagino1 & Takeshi Senga2 1Department of Surgical Oncology, Nagoya University Graduate School of Medicine, 65 Tsurumai, Showa, Nagoya 466-8550, Japan 2Division of Cancer Biology, Nagoya University Graduate School of Medicine, 65 Tsurumai, Showa, Nagoya 466-8550, Japan 3Division of Neurogenetics, Nagoya University Graduate School of Medicine, 65 Tsurumai, Showa, Nagoya 466-8550, Japan Keywords Abstract Apoptosis, EFTUD2, PRPF8, RNA splicing, SNRNP200, SNW1 RNA splicing is a fundamental process for protein synthesis. Recent studies have reported that drugs that inhibit splicing have cytotoxic effects on various Correspondence tumor cell lines. In this report, we demonstrate that depletion of SNW1, a Takeshi Senga, 65 Tsurumai, Showa, Nagoya component of the spliceosome, induces apoptosis in breast cancer cells. Proteo- 466-8550, Japan. Tel: 81-52-744-2463; mics and biochemical analyses revealed that SNW1 directly associates with Fax: 81-52-744-2464; other spliceosome components, including EFTUD2 (Snu114) and SNRNP200 E-mail: [email protected] (Brr2). The SKIP region of SNW1 interacted with the N-terminus of EFTUD2 Funding Information as well as two independent regions in the C-terminus of SNRNP200. Similar to This research was funded by a grant from SNW1 depletion, knockdown of EFTUD2 increased the numbers of apoptotic the Ministry of Education, Culture, Sports, cells. Furthermore, we demonstrate that exogenous expression of either the Science and Technology of Japan SKIP region of SNW1 or the N-terminus region of EFTUD2 significantly pro- (Nanomedicine molecular science, 23107010. -

Blueprint Genetics Craniosynostosis Panel
Craniosynostosis Panel Test code: MA2901 Is a 38 gene panel that includes assessment of non-coding variants. Is ideal for patients with craniosynostosis. About Craniosynostosis Craniosynostosis is defined as the premature fusion of one or more cranial sutures leading to secondary distortion of skull shape. It may result from a primary defect of ossification (primary craniosynostosis) or, more commonly, from a failure of brain growth (secondary craniosynostosis). Premature closure of the sutures (fibrous joints) causes the pressure inside of the head to increase and the skull or facial bones to change from a normal, symmetrical appearance resulting in skull deformities with a variable presentation. Craniosynostosis may occur in an isolated setting or as part of a syndrome with a variety of inheritance patterns and reccurrence risks. Craniosynostosis occurs in 1/2,200 live births. Availability 4 weeks Gene Set Description Genes in the Craniosynostosis Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ALPL Odontohypophosphatasia, Hypophosphatasia perinatal lethal, AD/AR 78 291 infantile, juvenile and adult forms ALX3 Frontonasal dysplasia type 1 AR 8 8 ALX4 Frontonasal dysplasia type 2, Parietal foramina AD/AR 15 24 BMP4 Microphthalmia, syndromic, Orofacial cleft AD 8 39 CDC45 Meier-Gorlin syndrome 7 AR 10 19 EDNRB Hirschsprung disease, ABCD syndrome, Waardenburg syndrome AD/AR 12 66 EFNB1 Craniofrontonasal dysplasia XL 28 116 ERF Craniosynostosis 4 AD 17 16 ESCO2 SC phocomelia syndrome, Roberts syndrome