Hypothalamic Glial Cells in Diet Induced Obesity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Motor Competence in Visually Impaired Persons
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/341870616 Motor competence in visually impaired persons Book · January 2019 DOI: 10.5507/ftk.19.24455549 CITATIONS READS 0 20 2 authors, including: Janečka Zbyněk Palacký University Olomouc 15 PUBLICATIONS 19 CITATIONS SEE PROFILE Some of the authors of this publication are also working on these related projects: dis HBSC - monitoring of lifestyle of students with special education needs View project Centra podpory integrace / Center of inclusion support View project All content following this page was uploaded by Janečka Zbyněk on 27 November 2020. The user has requested enhancement of the downloaded file. Palacký University Olomouc Faculty of Physical Culture Motor competence in visually impaired persons Zbyněk Janečka Ladislav Bláha et al. Olomouc 2019 Reviewers: Ken Black Maria Dinold First Edition Unauthorized use of the work is a breach of copyright and may be subject to civil, administrative or criminal liability. © Zbyněk Janečka, Ladislav Bláha, 2019 © Palacký University Olomouc, 2019 DOI: 10.5507/ftk.19.24455549 ISBN 978-80-244-5554-9 (print) ISBN 978-80-244-5555-6 (online : PDF) Content 1 / Terminology (Janečka, Bláha) ............................................................................................................7 2 / Type of impairment and its categorisation (Bláha) ..........................................................................11 3 / Vision (Janečka) .................................................................................................................................15 -

Preventive Aspects of Early Nutrition Nestlé Nutrition Institute Workshop Series
Preventive Aspects of Early Nutrition Nestlé Nutrition Institute Workshop Series Vol. 85 Preventive Aspects of Early Nutrition Editors Mary S. Fewtrell London, UK Ferdinand Haschke Salzburg, Austria Susan L. Prescott Perth, Australia Nestec Ltd., 55 Avenue Nestlé, CH–1800 Vevey (Switzerland) S. Karger AG, P.O. Box, CH–4009 Basel (Switzerland) www.karger.com Library of Congress Cataloging-in-Publication Data Names: Fewtrell, Mary S., editor. | Haschke, F., editor. | Prescott, Susan L., editor. Title: Preventive aspects of early nutrition / editors, Mary S. Fewtrell, Ferdinand Haschke, Susan L. Prescott. Description: Basel ; New York : Karger, [2016] | Series: Nestlé Nutrition Institute workshop series, ISSN 1664-2147 ; vol. 85 | Includes bibliographical references and index. Identifiers: LCCN 2015046033| ISBN 9783318056426 (hard cover : alk. paper) | ISBN 9783318056433 (electronic version) Subjects: | MESH: Child Nutritional Physiological Phenomena | Infant Nutritional Physiological Phenomena | Pediatric Obesity--prevention & control | Hypersensitivity--prevention & control | Congresses Classification: LCC RJ206 | NLM WS 130 | DDC 613.2083--dc23 LC record available at http://lccn.loc.gov/2015046033 The material contained in this volume was submitted as previously unpublished material, except in the instances in which credit has been given to the source from which some of the illustrative material was derived. Great care has been taken to maintain the accuracy of the information contained in the volume. However, neither Nestec Ltd. nor S. Karger AG can be held responsible for errors or for any consequences arising from the use of the information contained herein. © 2016 Nestec Ltd., Vevey (Switzerland) and S. Karger AG, Basel (Switzerland). All rights reserved. This book is protected by copyright. No part of it may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, or recording, or otherwise, without the written permission of the publisher. -

Annual Report 2008-2009
annual report 09 v4:Layout 1 11/8/2010 9:39 AM Page 1 Western Reserve Academy Annual Report 2008-2009 Behind each student and faculty member lie the loyalty, generosity and strength of the entire Western Reserve Academy community. annual report 09 v4:Layout 1 11/8/2010 9:39 AM Page 2 2008-2009 Board of Trustees Mark R. Tercek ’75, Daniel W. Christman ’61 Andrew R. Midler ’79 Douglas Kurdziel ’05, Co-president Alexandria, Va. St. Thomas, U.S. Virgin College Trustee Irvington, N.Y. Kathryn I. Clark ’76 Islands Nashville, Tenn. Peter S. Hellman ’68, Ann Arbor, Mich. Katie Ong-Landini ’83 Cookie Richardson, Co-president Ruth Swetland Eppig Shaker Heights, Ohio Pioneer Women President Hunting Valley, Ohio Bratenahl, Ohio Gregory Pennington ’71 Hudson, Ohio Stephan W. Cole ’66, Dagmar F. Fellowes ’75 Milwaukee, Wis. Lorraine Debose ’93, Vice President Pepper Pike, Ohio R. Lawrence Roth Alumni Association President Jupiter, Fla. New York, N.Y. Euclid, Ohio John M. Fowler ’67 Trustee Emeriti David M. Hunter ’68, Bronxville, N.Y. Hewitt B. Shaw Vice President Hudson, Ohio H. Arthur Bellows Jr. ’56 Silver Lake, Ohio Anne C. Goodman ’84 Shaker Heights, Ohio Shannah Tharp-Taylor ’91 Greenwich, Conn. Richard W. Neu, Ronald M. Harrington ’87 Pittsburgh, Pa. Jefferson W. Keener Jr. ’50 Treasurer Akron, Ohio Naples, Fla. Hudson, Ohio Jason M. Wortendyke ’94 Olin J. Heestand Jr. ’62 Chicago, Ill. T. Dixon Long ’51 Fred A. Cummings ’85, San Anselmo, Calif. Assistant Treasurer Chicago, Ill. Special Trustees Moreland Hills, Ohio Nathaniel E. Leonard ’82 David J. -

Lrtsv27no3.Pdf
EDITORIATBOARD Editor,andChairpersonoJtheEditoriatBoard. .. Er-rzesl.rlrL'Tare AssistantEditors Psvllrs A RrcnrroNo .for Cataloging and Classification Section Eow,A,noSwaNsoN Cenorvr C. Monnow for Presewation of Library Materials Section FneNcrsF. SpnErrznr for Reproduction of Library Materials Section Section J. Mrcueel Bnuen for Resources LrNor Sepp for Serials Section Editorial Adoisu: Donts H Cr-acx (for Regional Groups) Liaisonuith RTSD Newsletter: Atrqor-o HrnsnoN, RTSD NewsletterEditor Library ResourcesI TechnicalSerares (ISSN 0024-252?), the quarterly official publication ofthe Resourcesand Technical ServicesDivision of the American Library Association,is publishedat 50 E.HuronSt.,Chicago, IL60611. BusinessOffice:AmericanLibraryAssociation,50E.HuronSt., Chicago, IL 60611. Aduntising Tralfic Coordinator;Leona Swiech, Central Production Unit, ALA Headquarters,50E. HuronSt., Chicago, IL60611. CirculationandProduction:Central Production Unit{turnals, ALA Headquarters,50 E. Huron St., Chicago,IL 60611 . SubscriptionPrice. to mem- bersof the ALA Resourcesand Technical ServicesDivision, $10 per year, included in the member- ship dues; to nonmembers, $20 per year; singlecopies $5. Second-classpostage paid at Chicago, Illinois, and at additional mailing offices.POSTMAS- TER: Send addiess changes to Librarl ResourcesI TechnicalSeruices, 50 E. Huron St., Chicago, IL 6061I Librar2 ResourcesI TechnicalSeraices is indexed in Library Literature,Librarl I InformationScience Index- Con- Absmai, CurrentIndex toJournals in Education, ScienceCitation Index, and Hospital Literature tentsarelistedin CALL(CurrmtAuareness-Librar\Literature).ItsreviewsareincludedinBookRetieu Digest,Book Reouw Infux, and Retieu of Reuieus Copies of books for review should be addressedto Arnold Hirshon, Editor, R ZSD Newsletter,Ca- bell Library, virginia commonwealth ljniversity, 901 Park Ave., Richmond, VA 23284. Do not send journal issuesor journal articles for review. The contents of this journal, unless otheruise indicated, are copyrighted by the Association. -

Där Östersjön Är Västersjön Seal, Kus Läänemeri on Idameri
Där Östersjön är Västersjön Seal, kus Läänemeri on Idameri Där Östersjön är Västersjön Seal, kus Läänemeri on Idameri Festskrift till Virve och Raimo Raag Pühendusteos Virve ja Raimo Raagile ROGIER BLOKLAND OCH RIITTA-LIISA VALIJÄRVI (RED.) 2020 © The Authors 2020 Cover image: © Wikimedia Commons Production: Graphic Services, Uppsala University Distributed by Uppsala University, Department of Modern Languages, Box 636, 751 26 Uppsala, Sweden www.moderna.uu.se [email protected] ISBN 978-91-506-2835-7 http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-416455 Printed by Kph Trycksaksbolaget AB, Uppsala Tabula Gratulatoria Rogier Blokland, Uppsala Renate ja Karl Pajusalu, Tartu Tünde Blomqvist, Uppsala Helen Plado, Helsinki & Tartu Eesti Instituut, Tallinn & Võru Eesti Keele Instituut, Tallinn Aivar Põldvee, Tallinn Eesti Vabariigi Suursaatkond Eva Pärt Enander, Uppsala Stockholmis Nicolaus Janos Raag, Uppsala Märit Frändén, Uppsala Heiki Reila, Tallinn Johan Gille, Uppsala Thomas Rosén, Göteborg Thomas Grub, Uppsala Kristiina Ross, Tallinn Satu Gröndahl, Uppsala Meeli Sedrik, Tartu Riho Grünthal, Helsinki Dessislava Stoeva-Holm, Uppsala Anders Gustafsson, Uppsala Lars-Göran Sundell, Uppsala Cornelius Hasselblatt, Zuidwolde Torbjörn Söder, Uppsala Mervi de Heer, Uppsala Tiina Söderman, Tallinn Institutionen för moderna språk, Kai Tafenau, Tallinn Uppsala Laura Talvineva, Uppsala Olena Jansson, Uppsala Tartu Ülikooli eesti ja Leelo Keevallik, Linköping üldkeeleteaduse instituut, Tartu Olle Kejonen, Uppsala Tõnu Tender, Tallinn Katrin Kern, Tartu -

Kuje Li Se Sreća?
D a g l a s , Đesika, Li, Lukas, Nensy, Lennox, Noah, Sarah, Tomas, Bezuh, OdGrdan, Mile Grlica, doHlap, D rageKlempo, Koljibaba, Nedostoja, Poruga, Ukraden, Zlojutro, DomAgoJ VIDOVIć Kuje li se sreća? Uvod U mojemu je susjedstvu na Braču živio obrtnik koji ni u mirovini nije mirovao, a jačinu bih udaraca njegova mlata (tako su Hrvati nekoć nazivali čekić), koji su počinjali kad bi zora tek zarudjela, mjerio pomicanjem obrva nakon svakoga tupog odjeka udarca u predmet na nakovnju. Prisjetih se danas te priče iz davnine jer su na red za obradu došli predstavnici treće skupine prezimena po broju nositelja na hrvatskome ozemlju, skupine prezimena motiviranih nazivima zanimanja. Od njih su znatno brojnija prezimena nastala od osobnih imena (tu skupinu čine prezimena kao što su Jurić, Petrović, Radić) i prezimena nadimačkoga postanja (primjerice Babić, Bitanga, Nosek), a nešto su rjeđa prezimena etničkoga i etnonimskoga postanja (npr. Cetinski, Crnogorac, Slunjski, Vlašić). Prezimena su nastala od naziva zanimanja znatno češća u sjevernim hrvatskim krajevima, u kojima je obrt bio razvijeniji (npr. Klobučar ‘osoba koja izrađuje šešire’, Šustar ʻpostolarʼ, Špoljar ʻbravarʼ). Najčešća su prezimena iz navedene skupine motivirana nazivom kovač, te ćemo se u idućim redcima prošetati ne samo Hrvatskom nego i Europom kako bismo im ušli u trag. Kovači svi i svuda Naziv je kovač zajednički svim južnoslavenskim jezicima te zapadnoslavenskomu slovačkom jeziku (u tome se jeziku zapisuje kao kováč) i ugrofinskomu mađarskom jeziku (zapisuje se kovács), koji je navedeni naziv primio iz slavenskih jezika. Među govornicima svih spomenutih jezika te u prilagođenoj inačici u rumunjskome jeziku (Covaci) poprilično je velik broj nositelja prezimena Kovač neovisno o njegovu zapisu (prezime je Kováč drugo po čestoći u Slovačkoj, Kovács treće u Mađarskoj, a Kovač sedmo u Sloveniji). -
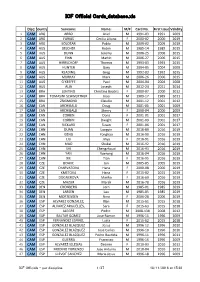
ICF Official Cards Database.Xls
ICF Official Cards_database.xls Disc:Country Surname: Name: M/F Card No. First Issue Validity 1 CAM ARG ARBO Ariel M 1991‐03 1991 2009 2 CAM ARG FARIAS Cecilia Liliana F 2000‐02 2000 2019 3 CAM ARG SOLOTAR Pablo M 2009‐02 2009 2019 4 CAM AUS BELCHER Robin M 1985‐14 1985 2015 5 CAM AUS DUNN Jeremy M 2006‐25 2006 2015 6 CAM AUS FINN Martin M 2006‐27 2006 2015 7 CAM AUS HIRRSCHOFF Thomas M 1993‐03 1993 2015 8 CAM AUS HUNTER Gary M 2004‐05 2004 2008 9 CAM AUS KEADING Greg M 1992‐03 1992 2015 10 CAM AUS MURRAY Mark M 2006‐26 2006 2015 11 CAM AUS O'KEEFFE Paul M 2004‐04 2004 2008 12 CAM AUS ALIA Joseph M 2012‐24 2011 2014 13 CAM BRA LENTINO Christina Beatriz F 2000‐07 2000 2012 14 CAM BRA TOMASINI SCHWERTNER Joao M 1989‐17 1989 2011 15 CAM BRA ZSIGMOND Claudio M 2001‐12 2001 2012 16 CAM CAN ARCHIBALD Doug M 2001‐05 2001 2009 17 CAM CAN ARCHIBALD Sherry F 2000‐04 2000 2009 18 CAM CAN CORBIN Doris F 2001‐01 2001 2017 19 CAM CAN CORBIN Dwight M 2001‐09 2001 2017 20 CAM CAN RICKARD Susan F 2001‐06 2001 2017 21 CAM CHN DUAN Longyin M 2016‐89 2016 2019 22 CAM CHN GENG Yonghao M 2016‐90 2016 2019 23 CAM CHN LIU Jinyu F 2016‐91 2016 2019 24 CAM CHN MAO Shukai M 2016‐92 2016 2019 25 CAM CHN SHI Chengzhouzi M 2016‐93 2016 2019 26 CAM CHN YAN Xuerong M 2016‐94 2016 2019 27 CAM CHN XIE Tian F 2016‐95 2016 2019 28 CAM CZE BOHAC Jan M 2005‐05 2005 2019 29 CAM CZE CHARVATOVA Hana F 2000‐08 2000 2019 30 CAM CZE KMETOVA Hana F 2015‐02 2015 2019 31 CAM CZE DOGNEROVA Marika F 2016‐69 2016 2019 32 CAM CZE MAZUR Marek M 2016‐70 2016 2019 33 CAM DEN CRONBERG Jorn -

Proceedings of the 7Th Workshop on Balto-Slavic Natural Language Processing, Pages 1–11, Florence, Italy, 2 August 2019
BSNLP’2019 The 7th Workshop on Balto-Slavic Natural Language Processing Proceedings of the Workshop BSNLP’2019 August 2, 2019 Florence, Italy c 2019 The Association for Computational Linguistics Order copies of this and other ACL proceedings from: Association for Computational Linguistics (ACL) 209 N. Eighth Street Stroudsburg, PA 18360 USA Tel: +1-570-476-8006 Fax: +1-570-476-0860 [email protected] ISBN 978-1-950737-41-3 ii Preface This volume contains the papers presented at BSNLP-2019: the Seventh Workshop on Balto-Slavic Natural Language Processing. The workshop is organized by ACL SIGSLAV—the Special Interest Group on NLP in Slavic Languages of the Association for Computational Linguistics. The BSNLP workshops have been convening for over a decade, with a clear vision and purpose. On one hand, the languages from the Balto-Slavic group play an important role due to their widespread use and diverse cultural heritage. These languages are spoken by about one-third of all speakers of the official languages of the European Union, and by over 400 million speakers worldwide. The political and economic developments in Central and Eastern Europe place societies where Balto-Slavic languages are spoken at the center of rapid technological advancement and growing European consumer markets. On the other hand, research on theoretical and applied NLP in some of these languages still lag behind the “major” languages, such as English and other Western European languages. In comparison to English, which has dominated the digital world since the advent of the Internet, many of these languages still lack resources, processing tools and applications—especially those with smaller speaker bases. -
Participant List
Participant List 10/20/2020 12:59:08 PM Category First Name Last Name Position Organization Nationality CSO Jamal Aazizi Chargé de la logistique Association Tazghart Morocco Luz Abayan Program Officer Child Rights Coalition Asia Philippines Babak Abbaszadeh President And Chief Toronto Centre For Global Canada Executive Officer Leadership In Financial Supervision Amr Abdallah Director, Gulf Programs Education for Employment - United States EFE Ziad Abdel Samad Executive Director Arab NGO Network for Lebanon Development TAZI Abdelilah Président Associaion Talassemtane pour Morocco l'environnement et le développement ATED Abla Abdellatif Executive Director and The Egyptian Center for Egypt Director of Research Economic Studies Nabil Abdo MENA Senior Policy Oxfam International Lebanon Advisor Baako Abdul-Fatawu Executive Director Centre for Capacity Ghana Improvement for the Wellbeing of the Vulnerable (CIWED) Maryati Abdullah Director/National Publish What You Pay Indonesia Coordinator Indonesia Dr. Abel Executive Director Reach The Youth Uganda Switzerland Mwebembezi (RTY) Suchith Abeyewickre Ethics Education Arigatou International Sri Lanka me Programme Coordinator Diam Abou Diab Fellow Arab NGO Network for Lebanon Development Hayk Abrahamyan Community Organizer for International Accountability Armenia South Caucasus and Project Central Asia Aliyu Abubakar Secretary General Kano State Peace and Conflict Nigeria Resolution Association Sunil Acharya Regional Advisor, Climate Practical Action Nepal and Resilience Salim Adam Public Health -
Author Index to Volume 56 Key to Abbreviations: (E) Editorial; (Er) Erratum; (GE) Guest Editorial; (L) Letter; (O) Obituary; (R) Review
European Journal of Clinical Nutrition (2002) 56, 1255–1258 ß 2002 Nature Publishing Group All rights reserved 0954–3007/02 $25.00 www.nature.com/ejcn Author Index to Volume 56 Key to abbreviations: (E) Editorial; (Er) Erratum; (GE) Guest Editorial; (L) Letter; (O) Obituary; (R) Review Abramson M 31 Bauer J 779 Bueno-de-Mesquita HB 1216 Damayanti K 1119 Abravicius JA Suppl 2 S18 Baye´sR44 Bugianesi R 546 Dammacco F 171 Abuja PM 72 Beard T 519 Burke V 1137 Dangour AD 1030 Accurso FJ 940 Becker N 1216 Burns AA 368 Dardenne M Suppl 3 S20 Adlercreutz H 157, 952 Becker W Suppl 2 S53, Burr ML 512 Davey G 1216 af Geijerstam A 221 Suppl 2 S75, Suppl 2 S89 Davies GJ 765 Agte VV 96 Bedogni G 1143 Cacciari E 171 Davies PSW 358, 650, 940 A˚ gren JJ 149 Behall KM 913 Caldara A 15 Day NE 882 Aitken W 748 Beitz R 539 Calder PC Suppl 3 S14 De Bosscher H 121 Akkila J 157 Bellussi L 1155 Calle AP 830 de Groot CPGM 245 Akuse RM 729 Benavente M 141 Campoy C 44 De Henauw S Suppl 2 S18, Al-Mousa Z 709 Benetou V 37 Campoy-Folgoso C 1242 Suppl 2 S53, Suppl 2 S75, Al-Raqua M 709 Bennett F 506 Cano P Suppl 3 S69 Suppl 2 S89 Albala C 200 Berglund G 1216 Capra S 779 De la Fuente M Suppl 3 S5 Albanes D 615 Berglund L 532 Carlsson E 680 de La Gue´ronnie`re V 442 Aldinucci C 1155 Bergsjø PB 192 Cashman KD 402 de la Rosa B Suppl 3 S57 Alfthan G 891 Bernhardt J 469 (R) Castellote AI 114 de la Torre-Boronat MC 114 Aller R 1126 Berthier AM 1011 Castellote-Bargallo´ AI 1242 De Leeuw I 121 Alme´ras N 297 Bertrais S 659 Cederholm T 221 De Luca F 171 Almdahl S 694 Bhan MK -

Warsaw School of Economics
ISSN 2081-9633 WWW.SGH.WAW.PL/WFES WFES Warsaw Forum of Economic Sociology Volume 2 Number 2(4) Autumn 2011 Warsaw School of Economics Collegium of Socio-Economics – Department of Economic Sociology © Copyright by Department of Economic Sociology, Collegium of Socio-Economics, Warsaw School of Economics, Warsaw 2012 All rights reserved. No part of this publication may be reproduced, stored or transmitted without the permission of the Warsaw School of Economics. First edition ISSN 2081-9633 Warsaw School of Economics – Publishing 02-554 Warszawa, al. Niepodległości 164 tel. +48-22 564 94 77, fax +48-22 564 86 86 www.wydawnictwo.sgh.waw.pl e-mail: [email protected] Printed by ESUS Drukarnia cyfrowa 62-835 Plewiska, ul. Południowa 54 Order 147/XI/12 Contents In this Issue ........................................................................................................................... 5 Vera Trappmann Labour as Entrepreneurs: Th e Ambivalent Role of Labour in Restructuring in the Polish Steel Industry .............................................................. 7 Sławomir Adamczyk and Barbara Surdykowska Can Trade Unions Contribute to the Future Competitiveness of the European Steel Industry? Th e Role of Social Dialogue in the Development of Qualifi cations and Competencies ..................................... 29 Rafał Towalski Social Dialogue as a Supporting Mechanism for Restructuring Processes....... 51 Leszek K. Gilejko Steel Industry: the Most Successful Example of the Sectoral Social Dialogue in Poland? ......................................................................................................... -

Isf World Schools Championship in Athletics
ISF WORLD SCHOOLS CHAMPIONSHIP IN ATHLETICS RESULTS 16th Jean Humbert Memorial, Split, 13.-19.5.2019 ISF WORLD SCHOOLS CHAMPIONSHIP IN ATHLETICS 16th Jean Humbert Memorial, Split, 13.-19.5.2019 15.5.2019 School Team Boys, shot put 5kg Group: 1 15.5.2019, 10:00 Pos. Bib Surname, name Born Team Result Points 1. 478 HUANG Wei-Lun 2002 DajHS 16,49 70 2. 259 ZULUETA Martin 2002 ColB 16,26 68 3. 442 ČOLAKOVIĆ Dušan 2001 ETŠMP 15,24 62 4. 307 LAPÈRE Thomas 2003 LBaLi 15,16 61 5. 37 BERTSCHLER Daniel 2001 HTLRa 14,68 58 6. 251 KOFFI David 2002 HBS 13,75 53 7. 350 ANAV Rom 2002 KSHOL 13,04 48 8. 225 CSIKÁN Jan Josef 2001 GTřin 12,88 47 9. 39 FRÖHLICH Clemens 2001 HTLRa 12,76 46 10. 374 BOLDANS Ralfs 2001 BVG 12,69 46 11. 154 ESCUDERO Juan 2003 VDiv 12,55 45 12. 375 EIZANS Lauris 2002 BVG 12,37 44 13. 198 MARTINIĆ Toni 2001 GVNST 12,12 42 14. 355 YAIR Nir 2001 KSHOL 11,86 41 15. 455 ŠIMUNE Kristian 2001 SššNi 10,70 34 16. 174 YONGCONG Li 2003 FoSHS 10,66 33 17. 14 KADI Toufik 2002 AlgB 9,89 28 18. 354 VANSO Neria 2002 KSHOL 7,86 17 19. 327 PATIDAR Dhananjay 2004 FKSSS 6,10 8 1st try 2nd try 3rd try 1. HUANG Wei-Lun 16,06 15,72 X 16,49 2. ZULUETA Martin 15,23 15,92 16,26 X 3. ČOLAKOVIĆ Dušan X 15,24 X X 4.