Molecular Characterization of Microbial and Fungal Communities on Dry-Aged Beef of Hanwoo Using Metagenomic Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Bacterial Communities in Container Habitats of Mosquitoes
Microb Ecol (2008) 56:593–603 DOI 10.1007/s00248-008-9379-6 ORIGINAL ARTICLE Diversity of Bacterial Communities in Container Habitats of Mosquitoes Loganathan Ponnusamy & Ning Xu & Gil Stav & Dawn M. Wesson & Coby Schal & Charles S. Apperson Received: 8 February 2008 /Accepted: 16 February 2008 /Published online: 29 March 2008 # Springer Science + Business Media, LLC 2008 Abstract We investigated the bacterial diversity of micro- tainers consisted mainly of undescribed species, and a bial communities in water-filled, human-made and natural phylogenetic analysis based on 16S rRNA sequences sug- container habitats of the mosquitoes Aedes aegypti and gested that species composition was independent of both Aedes albopictus in suburban landscapes of New Orleans, container type and the spatial distribution of containers. Louisiana in 2003. We collected water samples from three Comparative PCR-based, cultivation-independent rRNA sur- classes of containers, including tires (n=12), cemetery urns veys of microbial communities associated with mosquito (n=23), and miscellaneous containers that included two tree habitats can provide significant insight into community holes (n=19). Total genomic DNA was extracted from water organization and dynamics of bacterial species. samples, and 16S ribosomal DNA fragments (operational taxonomic units, OTUs) were amplified by PCR and separated by denaturing gradient gel electrophoresis (DGGE). Introduction The bacterial communities in containers represented diverse DGGE-DNA banding patterns that were not related to the The mosquitoes Aedes aegypti and Aedes albopictus class of container or to the local spatial distribution of develop in water-filled, human-made containers that are containers. Mean richness and evenness of OTUs were highest distributed in urban and suburban landscapes [16]. -

DNA Barcoding in <I>Mucorales</I>: an Inventory of Biodiversity
UvA-DARE (Digital Academic Repository) DNA barcoding in Mucorales: an inventory of biodiversity Walther, G.; Pawłowska, J.; Alastruey-Izquierdo, A.; Wrzosek, W.; Rodriguez-Tudela, J.L.; Dolatabadi, S.; Chakrabarti, A.; de Hoog, G.S. DOI 10.3767/003158513X665070 Publication date 2013 Document Version Final published version Published in Persoonia - Molecular Phylogeny and Evolution of Fungi Link to publication Citation for published version (APA): Walther, G., Pawłowska, J., Alastruey-Izquierdo, A., Wrzosek, W., Rodriguez-Tudela, J. L., Dolatabadi, S., Chakrabarti, A., & de Hoog, G. S. (2013). DNA barcoding in Mucorales: an inventory of biodiversity. Persoonia - Molecular Phylogeny and Evolution of Fungi, 30, 11-47. https://doi.org/10.3767/003158513X665070 General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:29 Sep 2021 Persoonia 30, 2013: 11–47 www.ingentaconnect.com/content/nhn/pimj RESEARCH ARTICLE http://dx.doi.org/10.3767/003158513X665070 DNA barcoding in Mucorales: an inventory of biodiversity G. -

Molecular Identification of Fungi
Molecular Identification of Fungi Youssuf Gherbawy l Kerstin Voigt Editors Molecular Identification of Fungi Editors Prof. Dr. Youssuf Gherbawy Dr. Kerstin Voigt South Valley University University of Jena Faculty of Science School of Biology and Pharmacy Department of Botany Institute of Microbiology 83523 Qena, Egypt Neugasse 25 [email protected] 07743 Jena, Germany [email protected] ISBN 978-3-642-05041-1 e-ISBN 978-3-642-05042-8 DOI 10.1007/978-3-642-05042-8 Springer Heidelberg Dordrecht London New York Library of Congress Control Number: 2009938949 # Springer-Verlag Berlin Heidelberg 2010 This work is subject to copyright. All rights are reserved, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilm or in any other way, and storage in data banks. Duplication of this publication or parts thereof is permitted only under the provisions of the German Copyright Law of September 9, 1965, in its current version, and permission for use must always be obtained from Springer. Violations are liable to prosecution under the German Copyright Law. The use of general descriptive names, registered names, trademarks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. Cover design: WMXDesign GmbH, Heidelberg, Germany, kindly supported by ‘leopardy.com’ Printed on acid-free paper Springer is part of Springer Science+Business Media (www.springer.com) Dedicated to Prof. Lajos Ferenczy (1930–2004) microbiologist, mycologist and member of the Hungarian Academy of Sciences, one of the most outstanding Hungarian biologists of the twentieth century Preface Fungi comprise a vast variety of microorganisms and are numerically among the most abundant eukaryotes on Earth’s biosphere. -

Redalyc.Mycotypha Indica P.M. Kirk & Benny, in Turkey Dung, a New Record
Multiciencias ISSN: 1317-2255 [email protected] Universidad del Zulia Venezuela Delgado Ávila, Adolfredo E.; Urdaneta García, Lilia M.; Piñeiro Chávez, Albino J. Mycotypha indica P.M. Kirk & Benny, in turkey dung, a new record for Venezuela Multiciencias, vol. 7, núm. 2, mayo-agosto, 2007, pp. 176-180 Universidad del Zulia Punto Fijo, Venezuela Available in: http://www.redalyc.org/articulo.oa?id=90470208 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative Ciencias del Agro y del Mar MULTICIENCIAS, Vol. 7, Nº 2, 2007 (176 - 180) ISSN 1317-2255 / Dep. legal pp. 200002FA828 Mycotypha indica P.M.Kirk & Benny, in turkey dung, a new record for Venezuela Adolfredo E. Delgado Ávila1, Lilia M. Urdaneta García1 y Albino J. Piñeiro Chávez1 1 Departamento Fitosanitario. Facultad de Agronomía. Universidad del Zulia. Apartado 526. Maracaibo ZU 4005. Venezuela. E-mail: [email protected], [email protected], [email protected] Abstract On the basis of a study of coprophilous fungi from Zulia state, Venezuela, a Mycotypha- ceae (Mucorales) Zygomycota with umbranched sporophores at first, often secondarily bran- ched; more or less erect, up to 3-4 mm high, 6-8 µm diam; hyaline at first, becoming pale blush gray, non-septate distally below the fertile vesicle. It is variable in length, ovoid to long-cylindri- cal minutely roughened; without sporangiola, rounded at apex, sporangiola dimorphic and borne in the outer row, are obvoid sporangiospores of similar size and shape to the sporangiola. -

Microorganisms in the Deterioration and Preservation of Cultural Heritage
Edith Joseph Editor Microorganisms in the Deterioration and Preservation of Cultural Heritage Microorganisms in the Deterioration and Preservation of Cultural Heritage Edith Joseph Editor Microorganisms in the Deterioration and Preservation of Cultural Heritage Editor Edith Joseph Institute of Chemistry University of Neuchâtel Neuchâtel, Switzerland Haute Ecole Arc Conservation Restauration University of Applied Sciences and Arts HES-SO Neuchâtel, Switzerland ISBN 978-3-030-69410-4 ISBN 978-3-030-69411-1 (eBook) https://doi.org/10.1007/978-3-030-69411-1 © The Editors(s) (if applicable) and The Author(s) 2021. This book is an open access publication. Open Access This book is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made. The images or other third party material in this book are included in the book's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the book's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. -

(12) United States Patent (10) Patent No.: US 7476,532 B2 Schneider Et Al
USOO7476532B2 (12) United States Patent (10) Patent No.: US 7476,532 B2 Schneider et al. (45) Date of Patent: Jan. 13, 2009 (54) MANNITOL INDUCED PROMOTER Makrides, S.C., "Strategies for achieving high-level expression of SYSTEMIS IN BACTERAL, HOST CELLS genes in Escherichia coli,” Microbiol. Rev. 60(3):512-538 (Sep. 1996). (75) Inventors: J. Carrie Schneider, San Diego, CA Sánchez-Romero, J., and De Lorenzo, V., "Genetic engineering of nonpathogenic Pseudomonas strains as biocatalysts for industrial (US); Bettina Rosner, San Diego, CA and environmental process.” in Manual of Industrial Microbiology (US) and Biotechnology, Demain, A, and Davies, J., eds. (ASM Press, Washington, D.C., 1999), pp. 460-474. (73) Assignee: Dow Global Technologies Inc., Schneider J.C., et al., “Auxotrophic markers pyrF and proC can Midland, MI (US) replace antibiotic markers on protein production plasmids in high cell-density Pseudomonas fluorescens fermentation.” Biotechnol. (*) Notice: Subject to any disclaimer, the term of this Prog., 21(2):343-8 (Mar.-Apr. 2005). patent is extended or adjusted under 35 Schweizer, H.P.. "Vectors to express foreign genes and techniques to U.S.C. 154(b) by 0 days. monitor gene expression in Pseudomonads. Curr: Opin. Biotechnol., 12(5):439-445 (Oct. 2001). (21) Appl. No.: 11/447,553 Slater, R., and Williams, R. “The expression of foreign DNA in bacteria.” in Molecular Biology and Biotechnology, Walker, J., and (22) Filed: Jun. 6, 2006 Rapley, R., eds. (The Royal Society of Chemistry, Cambridge, UK, 2000), pp. 125-154. (65) Prior Publication Data Stevens, R.C., “Design of high-throughput methods of protein pro duction for structural biology.” Structure, 8(9):R177-R185 (Sep. -

Antibiotic Resistant Pseudomonas Spp. Spoilers in Fresh Dairy Products: an Underestimated Risk and the Control Strategies
foods Review Antibiotic Resistant Pseudomonas Spp. Spoilers in Fresh Dairy Products: An Underestimated Risk and the Control Strategies Laura Quintieri , Francesca Fanelli * and Leonardo Caputo Institute of Sciences of Food Production, National Research Council of Italy, Via G. Amendola 122/O, 70126 Bari, Italy * Correspondence: [email protected]; Tel.: +39-0805929317 Received: 19 July 2019; Accepted: 23 August 2019; Published: 1 September 2019 Abstract: Microbial multidrug resistance (MDR) is a growing threat to public health mostly because it makes the fight against microorganisms that cause lethal infections ever less effective. Thus, the surveillance on MDR microorganisms has recently been strengthened, taking into account the control of antibiotic abuse as well as the mechanisms underlying the transfer of antibiotic genes (ARGs) among microbiota naturally occurring in the environment. Indeed, ARGs are not only confined to pathogenic bacteria, whose diffusion in the clinical field has aroused serious concerns, but are widespread in saprophytic bacterial communities such as those dominating the food industry. In particular, fresh dairy products can be considered a reservoir of Pseudomonas spp. resistome, potentially transmittable to consumers. Milk and fresh dairy cheeses products represent one of a few “hubs” where commensal or opportunistic pseudomonads frequently cohabit together with food microbiota and hazard pathogens even across their manufacturing processes. Pseudomonas spp., widely studied for food spoilage effects, are instead underestimated for their possible impact on human health. Recent evidences have highlighted that non-pathogenic pseudomonads strains (P. fluorescens, P. putida) are associated with some human diseases, but are still poorly considered in comparison to the pathogen P. aeruginosa. -

Shrimp Quality and Safety Management Along the Supply Chain in Benin
Shrimp quality and safety management along the supply chain in Benin D. Sylvain Dabadé Thesis committee Promotors Prof. Dr M.H. Zwietering Professor of Food Microbiology Wageningen University Prof. Dr D.J. Hounhouigan Professor of Food Science and Technology University of Abomey-Calavi, Benin Co-promotor Dr H.M.W. den Besten Assistant professor, Laboratory of Food Microbiology Wageningen University Other members Prof. Dr J.A.J. Verreth, Wageningen University Prof. Dr P. Dalgaard, Technical University of Denmark, Denmark Prof. Dr F. van Knapen, Utrecht University Dr E. Franz, National Institute for Public Health and the Environment, Bilthoven This research was conducted under the auspices of the Graduate School VLAG (Advanced studies in Food Technology, Agrobiotechnology, Nutrition and Health Sciences) Shrimp quality and safety management along the supply chain in Benin D. Sylvain Dabadé Thesis submitted in fulfilment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr A.P.J. Mol, in the presence of the Thesis committee appointed by the Academic Board to be defended in public on Tuesday 25 August 2015 at 11 a.m. in the Aula. D. Sylvain Dabadé Shrimp quality and safety management along the supply chain in Benin, 158 pages. PhD thesis, Wageningen University, Wageningen, NL (2015) With references, with summary in English ISBN 978-94-6257-420-5 Contents Abstract 7 Chapter 1 Introduction and outline of the thesis 9 Chapter 2 Quality perceptions of stakeholders in Beninese -

Deciphering the Diversity of Culturable Thermotolerant Bacteria from Manikaran Hot Springs
Ann Microbiol (2014) 64:741–751 DOI 10.1007/s13213-013-0709-7 ORIGINAL ARTICLE Deciphering the diversity of culturable thermotolerant bacteria from Manikaran hot springs Murugan Kumar & Ajar Nath Yadav & Rameshwar Tiwari & Radha Prasanna & Anil Kumar Saxena Received: 25 February 2013 /Accepted: 1 August 2013 /Published online: 24 August 2013 # Springer-Verlag Berlin Heidelberg and the University of Milan 2013 Abstract The aim of this study was to analyze and charac- Introduction terize the diversity of culturable thermotolerant bacteria in Manikaran hot springs. A total of 235 isolates were obtained Exotic niches, such as thermal springs, harbor populations of employing different media, and screened for temperature tol- microorganisms that can be a source of commercially impor- erance (40 °C–70 °C). A set of 85 isolates tolerant to 45 °C or tant products like enzymes, sugars, compatible solutes and above were placed in 42 phylogenetic clusters after amplified antibiotics (Satyanarayana et al. 2005). Thermal springs are a ribosomal DNA restriction analysis (16S rRNA-ARDRA). manifestation of geological activity and represent aquatic Sequencing of the 16S rRNA gene of 42 representative iso- microcosms that are produced by the emergence of geother- lates followed by BLAST search revealed that the majority of mally heated groundwater from the Earth’s crust. Prokaryotes isolates belonged to Firmicutes, followed by equal represen- are the major component of most ecosystems, being ubiqui- tation of Actinobacteria and Proteobacteria. Screening of rep- tous in nature because of their small size, easy dispersal, resentative isolates (42 ARDRA phylotypes) for amylase metabolic versatility, ability to utilize a broad range of nutri- activity revealed that 26 % of the isolates were positive, while ents, and tolerance to unfavorable and extreme conditions. -

Aquatic Microbial Ecology 80:15
The following supplement accompanies the article Isolates as models to study bacterial ecophysiology and biogeochemistry Åke Hagström*, Farooq Azam, Carlo Berg, Ulla Li Zweifel *Corresponding author: [email protected] Aquatic Microbial Ecology 80: 15–27 (2017) Supplementary Materials & Methods The bacteria characterized in this study were collected from sites at three different sea areas; the Northern Baltic Sea (63°30’N, 19°48’E), Northwest Mediterranean Sea (43°41'N, 7°19'E) and Southern California Bight (32°53'N, 117°15'W). Seawater was spread onto Zobell agar plates or marine agar plates (DIFCO) and incubated at in situ temperature. Colonies were picked and plate- purified before being frozen in liquid medium with 20% glycerol. The collection represents aerobic heterotrophic bacteria from pelagic waters. Bacteria were grown in media according to their physiological needs of salinity. Isolates from the Baltic Sea were grown on Zobell media (ZoBELL, 1941) (800 ml filtered seawater from the Baltic, 200 ml Milli-Q water, 5g Bacto-peptone, 1g Bacto-yeast extract). Isolates from the Mediterranean Sea and the Southern California Bight were grown on marine agar or marine broth (DIFCO laboratories). The optimal temperature for growth was determined by growing each isolate in 4ml of appropriate media at 5, 10, 15, 20, 25, 30, 35, 40, 45 and 50o C with gentle shaking. Growth was measured by an increase in absorbance at 550nm. Statistical analyses The influence of temperature, geographical origin and taxonomic affiliation on growth rates was assessed by a two-way analysis of variance (ANOVA) in R (http://www.r-project.org/) and the “car” package. -

Microbial and Mineralogical Characterizations of Soils Collected from the Deep Biosphere of the Former Homestake Gold Mine, South Dakota
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln US Department of Energy Publications U.S. Department of Energy 2010 Microbial and Mineralogical Characterizations of Soils Collected from the Deep Biosphere of the Former Homestake Gold Mine, South Dakota Gurdeep Rastogi South Dakota School of Mines and Technology Shariff Osman Lawrence Berkeley National Laboratory Ravi K. Kukkadapu Pacific Northwest National Laboratory, [email protected] Mark Engelhard Pacific Northwest National Laboratory Parag A. Vaishampayan California Institute of Technology See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/usdoepub Part of the Bioresource and Agricultural Engineering Commons Rastogi, Gurdeep; Osman, Shariff; Kukkadapu, Ravi K.; Engelhard, Mark; Vaishampayan, Parag A.; Andersen, Gary L.; and Sani, Rajesh K., "Microbial and Mineralogical Characterizations of Soils Collected from the Deep Biosphere of the Former Homestake Gold Mine, South Dakota" (2010). US Department of Energy Publications. 170. https://digitalcommons.unl.edu/usdoepub/170 This Article is brought to you for free and open access by the U.S. Department of Energy at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in US Department of Energy Publications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Gurdeep Rastogi, Shariff Osman, Ravi K. Kukkadapu, Mark Engelhard, Parag A. Vaishampayan, Gary L. Andersen, and Rajesh K. Sani This article is available at DigitalCommons@University of Nebraska - Lincoln: https://digitalcommons.unl.edu/ usdoepub/170 Microb Ecol (2010) 60:539–550 DOI 10.1007/s00248-010-9657-y SOIL MICROBIOLOGY Microbial and Mineralogical Characterizations of Soils Collected from the Deep Biosphere of the Former Homestake Gold Mine, South Dakota Gurdeep Rastogi & Shariff Osman & Ravi Kukkadapu & Mark Engelhard & Parag A. -
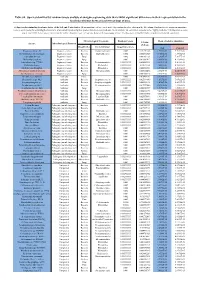
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp.