Endpoints in Vaccine Trials
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
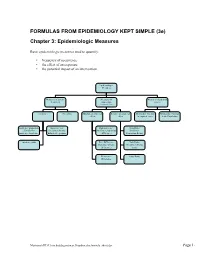
FORMULAS from EPIDEMIOLOGY KEPT SIMPLE (3E) Chapter 3: Epidemiologic Measures
FORMULAS FROM EPIDEMIOLOGY KEPT SIMPLE (3e) Chapter 3: Epidemiologic Measures Basic epidemiologic measures used to quantify: • frequency of occurrence • the effect of an exposure • the potential impact of an intervention. Epidemiologic Measures Measures of disease Measures of Measures of potential frequency association impact (“Measures of Effect”) Incidence Prevalence Absolute measures of Relative measures of Attributable Fraction Attributable Fraction effect effect in exposed cases in the Population Incidence proportion Incidence rate Risk difference Risk Ratio (Cumulative (incidence density, (Incidence proportion (Incidence Incidence, Incidence hazard rate, person- difference) Proportion Ratio) Risk) time rate) Incidence odds Rate Difference Rate Ratio (Incidence density (Incidence density difference) ratio) Prevalence Odds Ratio Difference Macintosh HD:Users:buddygerstman:Dropbox:eks:formula_sheet.doc Page 1 of 7 3.1 Measures of Disease Frequency No. of onsets Incidence Proportion = No. at risk at beginning of follow-up • Also called risk, average risk, and cumulative incidence. • Can be measured in cohorts (closed populations) only. • Requires follow-up of individuals. No. of onsets Incidence Rate = ∑person-time • Also called incidence density and average hazard. • When disease is rare (incidence proportion < 5%), incidence rate ≈ incidence proportion. • In cohorts (closed populations), it is best to sum individual person-time longitudinally. It can also be estimated as Σperson-time ≈ (average population size) × (duration of follow-up). Actuarial adjustments may be needed when the disease outcome is not rare. • In an open populations, Σperson-time ≈ (average population size) × (duration of follow-up). Examples of incidence rates in open populations include: births Crude birth rate (per m) = × m mid-year population size deaths Crude mortality rate (per m) = × m mid-year population size deaths < 1 year of age Infant mortality rate (per m) = × m live births No. -

Facilitating the Use of Imaging Biomarkers in Therapeutic Clinical
Facilitating the Use of Imaging Biomarkers in Therapeutic Clinical Trials Michael Graham, PhD, MD President, SNM Co-chair, Clinical Trials Network Facilitating the Use of Imaging Biomarkers in Therapeutic Clinical Trials • Definitions – Biomarker, Surrogate Biomarker • Standardization • Harmonization • Elements of a clinical trial • What can be facilitated • SNM Clinical Trials Network Imaging Biomarkers A biomarker is a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention. (FDA website) • Utility of imaging biomarkers in clinical trials – Assessing response to therapy (surrogate end point) • FDG • FLT – Stratifying patient populations • Receptor status (FES, SRS, etc.) • Hypoxia Surrogate Endpoints in Clinical Trials A surrogate endpoint is expected to predict clinical benefit (or harm, or lack of benefit) based on epidemiologic, therapeutic, pathophysiologic or other scientific evidence. (FDA website) • Assessing response to therapy – Relatively early “go vs. no go” decisions in Phase I or II – Decision point in adaptive designed trials – Building evidence for “validation” or “qualification” • Personalized medicine – Early identification of responders and non-responders Sohn HJ, et al. FLT PET before and 7 days after gefitinib (EGFR inhibitor) treatment predicts response in patients with advanced adenocarcinoma of the lung. Clin Cancer Res. 2008 Nov 15;14(22):7423-9. Imaging at 1 hr p 15 mCi FLT Threshold: -

Statistical Guidance on Reporting Results from Studies Evaluating Diagnostic Tests Document Issued On: March 13, 2007
Guidance for Industry and FDA Staff Statistical Guidance on Reporting Results from Studies Evaluating Diagnostic Tests Document issued on: March 13, 2007 The draft of this document was issued on March 12, 2003. For questions regarding this document, contact Kristen Meier at 240-276-3060, or send an e-mail to [email protected]. U.S. Department of Health and Human Services Food and Drug Administration Center for Devices and Radiological Health Diagnostic Devices Branch Division of Biostatistics Office of Surveillance and Biometrics Contains Nonbinding Recommendations Preface Public Comment Written comments and suggestions may be submitted at any time for Agency consideration to the Division of Dockets Management, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD, 20852. Alternatively, electronic comments may be submitted to http://www.fda.gov/dockets/ecomments. When submitting comments, please refer to Docket No. 2003D-0044. Comments may not be acted upon by the Agency until the document is next revised or updated. Additional Copies Additional copies are available from the Internet at: http://www.fda.gov/cdrh/osb/guidance/1620.pdf. You may also send an e-mail request to [email protected] to receive an electronic copy of the guidance or send a fax request to 240-276-3151 to receive a hard copy. Please use the document number 1620 to identify the guidance you are requesting. Contains Nonbinding Recommendations Table of Contents 1. Background....................................................................................................4 -

Estimated HIV Incidence and Prevalence in the United States
Volume 26, Number 1 Estimated HIV Incidence and Prevalence in the United States, 2015–2019 This issue of the HIV Surveillance Supplemental Report is published by the Division of HIV/AIDS Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, Centers for Disease Control and Prevention (CDC), U.S. Department of Health and Human Services, Atlanta, Georgia. Estimates are presented for the incidence and prevalence of HIV infection among adults and adolescents (aged 13 years and older) based on data reported to CDC through December 2020. The HIV Surveillance Supplemental Report is not copyrighted and may be used and reproduced without permission. Citation of the source is, however, appreciated. Suggested citation Centers for Disease Control and Prevention. Estimated HIV incidence and prevalence in the United States, 2015–2019. HIV Surveillance Supplemental Report 2021;26(No. 1). http://www.cdc.gov/ hiv/library/reports/hiv-surveillance.html. Published May 2021. Accessed [date]. On the Web: http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html Confidential information, referrals, and educational material on HIV infection CDC-INFO 1-800-232-4636 (in English, en Español) 1-888-232-6348 (TTY) http://wwwn.cdc.gov/dcs/ContactUs/Form Acknowledgments Publication of this report was made possible by the contributions of the state and territorial health departments and the HIV surveillance programs that provided surveillance data to CDC. This report was prepared by the following staff and contractors of the Division of HIV/AIDS Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, CDC: Laurie Linley, Anna Satcher Johnson, Ruiguang Song, Sherry Hu, Baohua Wu, H. -

Understanding Relative Risk, Odds Ratio, and Related Terms: As Simple As It Can Get Chittaranjan Andrade, MD
Understanding Relative Risk, Odds Ratio, and Related Terms: As Simple as It Can Get Chittaranjan Andrade, MD Each month in his online Introduction column, Dr Andrade Many research papers present findings as odds ratios (ORs) and considers theoretical and relative risks (RRs) as measures of effect size for categorical outcomes. practical ideas in clinical Whereas these and related terms have been well explained in many psychopharmacology articles,1–5 this article presents a version, with examples, that is meant with a view to update the knowledge and skills to be both simple and practical. Readers may note that the explanations of medical practitioners and examples provided apply mostly to randomized controlled trials who treat patients with (RCTs), cohort studies, and case-control studies. Nevertheless, similar psychiatric conditions. principles operate when these concepts are applied in epidemiologic Department of Psychopharmacology, National Institute research. Whereas the terms may be applied slightly differently in of Mental Health and Neurosciences, Bangalore, India different explanatory texts, the general principles are the same. ([email protected]). ABSTRACT Clinical Situation Risk, and related measures of effect size (for Consider a hypothetical RCT in which 76 depressed patients were categorical outcomes) such as relative risks and randomly assigned to receive either venlafaxine (n = 40) or placebo odds ratios, are frequently presented in research (n = 36) for 8 weeks. During the trial, new-onset sexual dysfunction articles. Not all readers know how these statistics was identified in 8 patients treated with venlafaxine and in 3 patients are derived and interpreted, nor are all readers treated with placebo. These results are presented in Table 1. -

Surrogate Endpoint Biomarkers for Cervical Cancer Chemoprevention Trials
Journal of Cellular Biochemistry, Supplement 23:113-124 (1 995) Surrogate Endpoint Biomarkers for Cervical Cancer Chemoprevention Trials Mack T. Ruffin IV, MD, MPH: Mohammed S. Qgaily, MD: Carolyn M. Johnston, MD? Lucie Gregoire, PhD: Wayne D. Lancaster, PhD; and Dean E. Brenner, MD6 Department of Family Practice, University of Michigan Medical Center, Ann Arbor, MI 48109-0708 Department of Internal Medicine, Division of Hematology and Oncology, Simpson Memorial Institute, Ann Arbor, MI 48109-0724 Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, Ann Arbor, MI 48109-0718 Department of Obstetrics and Gynecology, Wayne State University School of Medicine, Detroit, MI 48201 Department of Obstetrics and Gynecology, Center for Molecular Medicine and Genetics, Detroit, MI 48201 Department of Internal Medicine, Division of Hematology and Oncology, Simpson Memorial Institute, Ann Arbor, MI 48109-0724 Abstract Cervical intraepithelial neoplasia (CIN) represents a spectrum of epithelial changes that provide an excellent model for developing chemopreventive interventions for cervical cancer. Possible drug effect surrogate endpoint biomarkers are dependent on the agent under investigation. Published and preliminary clinical reports suggest retinoids and carotenoids are effective chemopreventive agents for CIN. Determination of plasma and tissue pharmacology of these agents and their metabolites could serve as drug effect intermediate endpoints. In addition, retinoic acid receptors could serve as both drug and biological effect intermediate endpoints. Possible biological effect surrogate endpoint biomarkers include cytomorphological parameters, proliferation markers, genomic markers, regulatory markers, and differentiation. Given the demonstrated causality of human papillomavirus (HPV) for cervical cancer, establishing the relationship to HPV will be an essential component of any biological intermediate endpoint biomarker. -

Outcome Reporting Bias in COVID-19 Mrna Vaccine Clinical Trials
medicina Perspective Outcome Reporting Bias in COVID-19 mRNA Vaccine Clinical Trials Ronald B. Brown School of Public Health and Health Systems, University of Waterloo, Waterloo, ON N2L3G1, Canada; [email protected] Abstract: Relative risk reduction and absolute risk reduction measures in the evaluation of clinical trial data are poorly understood by health professionals and the public. The absence of reported absolute risk reduction in COVID-19 vaccine clinical trials can lead to outcome reporting bias that affects the interpretation of vaccine efficacy. The present article uses clinical epidemiologic tools to critically appraise reports of efficacy in Pfzier/BioNTech and Moderna COVID-19 mRNA vaccine clinical trials. Based on data reported by the manufacturer for Pfzier/BioNTech vaccine BNT162b2, this critical appraisal shows: relative risk reduction, 95.1%; 95% CI, 90.0% to 97.6%; p = 0.016; absolute risk reduction, 0.7%; 95% CI, 0.59% to 0.83%; p < 0.000. For the Moderna vaccine mRNA-1273, the appraisal shows: relative risk reduction, 94.1%; 95% CI, 89.1% to 96.8%; p = 0.004; absolute risk reduction, 1.1%; 95% CI, 0.97% to 1.32%; p < 0.000. Unreported absolute risk reduction measures of 0.7% and 1.1% for the Pfzier/BioNTech and Moderna vaccines, respectively, are very much lower than the reported relative risk reduction measures. Reporting absolute risk reduction measures is essential to prevent outcome reporting bias in evaluation of COVID-19 vaccine efficacy. Keywords: mRNA vaccine; COVID-19 vaccine; vaccine efficacy; relative risk reduction; absolute risk reduction; number needed to vaccinate; outcome reporting bias; clinical epidemiology; critical appraisal; evidence-based medicine Citation: Brown, R.B. -

Ethics of Vaccine Research
COMMENTARY Ethics of vaccine research Christine Grady Vaccination has attracted controversy at every stage of its development and use. Ethical debates should consider its basic goal, which is to benefit the community at large rather than the individual. accines truly represent one of the mira- include value, validity, fair subject selection, the context in which it will be used and Vcles of modern science. Responsible for favorable risk/benefit ratio, independent acceptable to those who will use it. This reducing morbidity and mortality from sev- review, informed consent and respect for assessment considers details about the pub- eral formidable diseases, vaccines have made enrolled participants. Applying these princi- lic health need (such as the prevalence, bur- substantial contributions to global public ples to vaccine research allows consideration den and natural history of the disease, as health. Generally very safe and effective, vac- of some of the particular challenges inherent well as existing strategies to prevent or con- cines are also an efficient and cost-effective in testing vaccines (Box 1). trol it), the scientific data and possibilities way of preventing disease. Yet, despite their Ethically salient features of clinical vac- (preclinical and clinical data, expected brilliant successes, vaccines have always been cine research include the fact that it involves mechanism of action and immune corre- controversial. Concerns about the safety and healthy subjects, often (or ultimately) chil- lates) and the likely use of the vaccine (who untoward effects of vaccines, about disturb- dren and usually (at least when testing effi- will use and benefit from it, safety, cost, dis- ing the natural order, about compelling indi- cacy) in very large numbers. -

Disease Incidence, Prevalence and Disability
Part 3 Disease incidence, prevalence and disability 9. How many people become sick each year? 28 10. Cancer incidence by site and region 29 11. How many people are sick at any given time? 31 12. Prevalence of moderate and severe disability 31 13. Leading causes of years lost due to disability in 2004 36 World Health Organization 9. How many people become sick each such as diarrhoeal disease or malaria, it is common year? for individuals to be infected repeatedly and have several episodes. For such conditions, the number The “incidence” of a condition is the number of new given in the table is the number of disease episodes, cases in a period of time – usually one year (Table 5). rather than the number of individuals affected. For most conditions in this table, the figure given is It is important to remember that the incidence of the number of individuals who developed the illness a disease or condition measures how many people or problem in 2004. However, for some conditions, are affected by it for the first time over a period of Table 5: Incidence (millions) of selected conditions by WHO region, 2004 Eastern The Mediter- South- Western World Africa Americas ranean Europe East Asia Pacific Tuberculosisa 7.8 1.4 0.4 0.6 0.6 2.8 2.1 HIV infectiona 2.8 1.9 0.2 0.1 0.2 0.2 0.1 Diarrhoeal diseaseb 4 620.4 912.9 543.1 424.9 207.1 1 276.5 1 255.9 Pertussisb 18.4 5.2 1.2 1.6 0.7 7.5 2.1 Measlesa 27.1 5.3 0.0e 1.0 0.2 17.4 3.3 Tetanusa 0.3 0.1 0.0 0.1 0.0 0.1 0.0 Meningitisb 0.7 0.3 0.1 0.1 0.0 0.2 0.1 Malariab 241.3 203.9 2.9 8.6 0.0 23.3 2.7 -

COVID-19 Vaccine Trials
COVID-19 VACCINE TRIALS SUMMARY OF PFIZER AND MODERNA COVID-19 VACCINE RESULTS Some of the most common concerns voiced involve the A review of unblinded reactogenicity data from the speed with which these vaccines were developed and final analysis which consisted of a randomized subset whether they are safe or not. How were the companies of at least 8,000 participants 18 years and older in able to get these vaccines developed and ready for the phase 2/3 study demonstrates that the vaccine distribution so fast? Were they tested in persons who was well tolerated, with most solicited adverse events are most vulnerable? Are the vaccines safe? resolving shortly after vaccination. Even though COVID-19 vaccines were developed at While the vaccine was well tolerated overall, and side an extraordinary speed, companies were required effects lasted for only a day or two, persons taking to take all of the regulatory and operational steps the vaccine should be aware that the side effects normally required for all vaccine trials, so none of are more than is seen in general for the flu vaccine these steps were skipped. What did occur that does and be prepared for them. Most side effects were not normally occur was some upfront financial mild to moderate and included injection-site pain, assistance from the federal government (Moderna redness and swelling at the injection site, fatigue/ accepted, Pfizer declined) and federal regulatory tiredness, headache, chills, muscle pain, and joint agencies working closely with the companies, pain. “Severe” side effects, defined as those that providing near real-time data with companies prevent persons from continuing daily activities were receiving review and advice more quickly. -

Adaptive Designs in Clinical Trials: Why Use Them, and How to Run and Report Them Philip Pallmann1* , Alun W
Pallmann et al. BMC Medicine (2018) 16:29 https://doi.org/10.1186/s12916-018-1017-7 CORRESPONDENCE Open Access Adaptive designs in clinical trials: why use them, and how to run and report them Philip Pallmann1* , Alun W. Bedding2, Babak Choodari-Oskooei3, Munyaradzi Dimairo4,LauraFlight5, Lisa V. Hampson1,6, Jane Holmes7, Adrian P. Mander8, Lang’o Odondi7, Matthew R. Sydes3,SofíaS.Villar8, James M. S. Wason8,9, Christopher J. Weir10, Graham M. Wheeler8,11, Christina Yap12 and Thomas Jaki1 Abstract Adaptive designs can make clinical trials more flexible by utilising results accumulating in the trial to modify the trial’s course in accordance with pre-specified rules. Trials with an adaptive design are often more efficient, informative and ethical than trials with a traditional fixed design since they often make better use of resources such as time and money, and might require fewer participants. Adaptive designs can be applied across all phases of clinical research, from early-phase dose escalation to confirmatory trials. The pace of the uptake of adaptive designs in clinical research, however, has remained well behind that of the statistical literature introducing new methods and highlighting their potential advantages. We speculate that one factor contributing to this is that the full range of adaptations available to trial designs, as well as their goals, advantages and limitations, remains unfamiliar to many parts of the clinical community. Additionally, the term adaptive design has been misleadingly used as an all-encompassing label to refer to certain methods that could be deemed controversial or that have been inadequately implemented. We believe that even if the planning and analysis of a trial is undertaken by an expert statistician, it is essential that the investigators understand the implications of using an adaptive design, for example, what the practical challenges are, what can (and cannot) be inferred from the results of such a trial, and how to report and communicate the results. -

Plant-Based COVID-19 Vaccines: Current Status, Design, and Development Strategies of Candidate Vaccines
Review Plant-Based COVID-19 Vaccines: Current Status, Design, and Development Strategies of Candidate Vaccines Puna Maya Maharjan 1 and Sunghwa Choe 2,3,* 1 G+FLAS Life Sciences, 123 Uiryodanji-gil, Osong-eup, Heungdeok-gu, Cheongju-si 28161, Korea; punamaya.maharjan@gflas.com 2 G+FLAS Life Sciences, 38 Nakseongdae-ro, Gwanak-gu, Seoul 08790, Korea 3 School of Biological Sciences, College of Natural Sciences, Seoul National University, Gwanak-gu, Seoul 08826, Korea * Correspondence: [email protected] Abstract: The prevalence of the coronavirus disease 2019 (COVID-19) pandemic in its second year has led to massive global human and economic losses. The high transmission rate and the emergence of diverse SARS-CoV-2 variants demand rapid and effective approaches to preventing the spread, diagnosing on time, and treating affected people. Several COVID-19 vaccines are being developed using different production systems, including plants, which promises the production of cheap, safe, stable, and effective vaccines. The potential of a plant-based system for rapid production at a commercial scale and for a quick response to an infectious disease outbreak has been demonstrated by the marketing of carrot-cell-produced taliglucerase alfa (Elelyso) for Gaucher disease and tobacco- produced monoclonal antibodies (ZMapp) for the 2014 Ebola outbreak. Currently, two plant-based COVID-19 vaccine candidates, coronavirus virus-like particle (CoVLP) and Kentucky Bioprocessing (KBP)-201, are in clinical trials, and many more are in the preclinical stage. Interim phase 2 clinical Citation: Maharjan, P.M.; Choe, S. trial results have revealed the high safety and efficacy of the CoVLP vaccine, with 10 times more Plant-Based COVID-19 Vaccines: neutralizing antibody responses compared to those present in a convalescent patient’s plasma.