Diatomic Vibrational Spectra
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
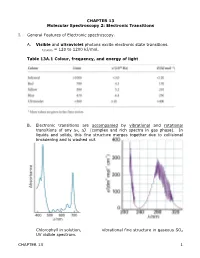
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions I. General Features of Electronic spectroscopy. A. Visible and ultraviolet photons excite electronic state transitions. εphoton = 120 to 1200 kJ/mol. Table 13A.1 Colour, frequency, and energy of light B. Electronic transitions are accompanied by vibrational and rotational transitions of any Δν, ΔJ (complex and rich spectra in gas phase). In liquids and solids, this fine structure merges together due to collisional broadening and is washed out. Chlorophyll in solution, vibrational fine structure in gaseous SO2 UV visible spectrum. CHAPTER 13 1 C. Molecules need not have permanent dipole to undergo electronic transitions. All that is required is a redistribution of electronic and nuclear charge between the initial and final electronic state. Transition intensity ~ |µ|2 where µ is the magnitude of the transition dipole moment given by: ˆ d µ = ∫ ψfinalµψ initial τ where µˆ = −e ∑ri + e ∑Ziri € electrons nuclei r vector position of the particle i = D. Franck-Condon principle: Because nuclei are so much more massive and € sluggish than electrons, electronic transitions can happen much faster than the nuclei can respond. Electronic transitions occur vertically on energy diagram at right. (Hence the name vertical transition) Transition probability depends on vibrational wave function overlap (Franck-Condon factor). In this example: ν = 0 ν’ = 0 have small overlap ν = 0 ν’ = 2 have greatest overlap Now because the total molecular wavefunction can be approximately factored into 2 terms, electronic wavefunction and the nuclear position wavefunction (using Born-Oppenheimer approx.) the transition dipole can also be factored into 2 terms also: S µ = µelectronic i,f S d Franck Condon factor i,j i,f = ∫ ψf,vibr ψi,vibr τ = − CHAPTER 13 2 See derivation “Justification 14.2” II. -

Lecture B4 Vibrational Spectroscopy, Part 2 Quantum Mechanical SHO
Lecture B4 Vibrational Spectroscopy, Part 2 Quantum Mechanical SHO. WHEN we solve the Schrödinger equation, we always obtain two things: 1. a set of eigenstates, |ψn>. 2. a set of eigenstate energies, En QM predicts the existence of discrete, evenly spaced, vibrational energy levels for the SHO. n = 0,1,2,3... For the ground state (n=0), E = ½hν. Notes: This is called the zero point energy. Optical selection rule -- SHO can absorb or emit light with a ∆n = ±1 The IR absorption spectrum for a diatomic molecule, such as HCl: diatomic Optical selection rule 1 -- SHO can absorb or emit light with a ∆n = ±1 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion. (Note: μ here means dipole moment). If a more realistic Morse potential is used in the Schrödinger Equation, these energy levels get scrunched together... ΔE=hν ΔE<hν The vibrational spectroscopy of polyatomic molecules gets more interesting... For diatomic or linear molecules: 3N-5 modes For nonlinear molecules: 3N-6 modes N = number of atoms in molecule The vibrational spectroscopy of polyatomic molecules gets more interesting... Optical selection rule 1 -- SHO can absorb or emit light with a ∆n = ±1 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion of a mode. Consider H2O (a nonlinear molecule): 3N-6 = 3(3)-6 = 3 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion of a mode. Consider H2O (a nonlinear molecule): 3N-6 = 3(3)-6 = 3 All bands are observed in the IR spectrum. -

2. Molecular Stucture/Basic Spectroscopy the Electromagnetic Spectrum
2. Molecular stucture/Basic spectroscopy The electromagnetic spectrum Spectral region fooatocadr atomic and molecular spectroscopy E. Hecht (2nd Ed.) Optics, Addison-Wesley Publishing Company,1987 Per-Erik Bengtsson Spectral regions Mo lecu lar spec troscopy o ften dea ls w ith ra dia tion in the ultraviolet (UV), visible, and infrared (IR) spectltral reg ions. • The visible region is from 400 nm – 700 nm • The ultraviolet region is below 400 nm • The infrared region is above 700 nm. 400 nm 500 nm 600 nm 700 nm Spectroscopy: That part of science which uses emission and/or absorption of radiation to deduce atomic/molecular properties Per-Erik Bengtsson Some basics about spectroscopy E = Energy difference = c /c h = Planck's constant, 6.63 10-34 Js ergy nn = Frequency E hn = h/hc /l E = h = hc / c = Velocity of light, 3.0 108 m/s = Wavelength 0 Often the wave number, , is used to express energy. The unit is cm-1. = E / hc = 1/ Example The energy difference between two states in the OH-molecule is 35714 cm-1. Which wavelength is needed to excite the molecule? Answer = 1/ =35714 cm -1 = 1/ = 280 nm. Other ways of expressing this energy: E = hc/ = 656.5 10-19 J E / h = c/ = 9.7 1014 Hz Per-Erik Bengtsson Species in combustion Combustion involves a large number of species Atoms oxygen (O), hydrogen (H), etc. formed by dissociation at high temperatures Diatomic molecules nitrogen (N2), oxygen (O2) carbon monoxide (CO), hydrogen (H2) nitr icoxide (NO), hy droxy l (OH), CH, e tc. -

Lecture 17 Highlights
Lecture 23 Highlights The phenomenon of Rabi oscillations is very different from our everyday experience of how macroscopic objects (i.e. those made up of many atoms) absorb electromagnetic radiation. When illuminated with light (like French fries at McDonalds) objects tend to steadily absorb the light and heat up to a steady state temperature, showing no signs of oscillation with time. We did a calculation of the transition probability of an atom illuminated with a broad spectrum of light and found that the probability increased linearly with time, leading to a constant transition rate. This difference comes about because the many absorption processes at different frequencies produce a ‘smeared-out’ response that destroys the coherent Rabi oscillations and results in a classical incoherent absorption. Selection Rules All of the transition probabilities are proportional to “dipole matrix elements,” such as x jn above. These are similar to dipole moment calculations for a charge distribution in classical physics, except that they involve the charge distribution in two different states, connected by the dipole operator for the transition. In many cases these integrals are zero because of symmetries of the associated wavefunctions. This gives rise to selection rules for possible transitions under the dipole approximation (atom size much smaller than the electromagnetic wave wavelength). For example, consider the matrix element z between states of the n,l , m ; n ', l ', m ' Hydrogen atom labeled by the quantum numbers n,,l m and n',','l m : z = ψ()()r z ψ r d3 r n,l , m ; n ', l ', m ' ∫∫∫ n,,l m n',l ', m ' To make progress, first consider just the φ integral. -

Introduction to Differential Equations
Quantum Dynamics – Quick View Concepts of primary interest: The Time-Dependent Schrödinger Equation Probability Density and Mixed States Selection Rules Transition Rates: The Golden Rule Sample Problem Discussions: Tools of the Trade Appendix: Classical E-M Radiation POSSIBLE ADDITIONS: After qualitative section, do the two state system, and then first and second order transitions (follow Fitzpatrick). Chain together the dipole rules to get l = 2,0,-2 and m = -2, -2, … , 2. ??Where do we get magnetic rules? Look at the canonical momentum and the Asquared term. Schrödinger, Erwin (1887-1961) Austrian physicist who invented wave mechanics in 1926. Wave mechanics was a formulation of quantum mechanics independent of Heisenberg's matrix mechanics. Like matrix mechanics, wave mechanics mathematically described the behavior of electrons and atoms. The central equation of wave mechanics is now known as the Schrödinger equation. Solutions to the equation provide probability densities and energy levels of systems. The time-dependent form of the equation describes the dynamics of quantum systems. http://scienceworld.wolfram.com/biography/Schroedinger.html © 1996-2006 Eric W. Weisstein www-history.mcs.st-andrews.ac.uk/Biographies/Schrodinger.html Quantum Dynamics: A Qualitative Introduction Introductory quantum mechanics focuses on time-independent problems leaving Contact: [email protected] dynamics to be discussed in the second term. Energy eigenstates are characterized by probability density distributions that are time-independent (static). There are examples of time-dependent behavior that are by demonstrated by rather simple introductory problems. In the case of a particle in an infinite well with the range [ 0 < x < a], the mixed state below exhibits time-dependence. -

Ijmp.Jor.Br V
INDEPENDENT JOURNAL OF MANAGEMENT & PRODUCTION (IJM&P) http://www.ijmp.jor.br v. 10, n. 8, Special Edition Seng 2019 ISSN: 2236-269X DOI: 10.14807/ijmp.v10i8.1046 A NEW HYPOTHESIS ABOUT THE NUCLEAR HYDROGEN STRUCTURE Relly Victoria Virgil Petrescu IFToMM, Romania E-mail: [email protected] Raffaella Aversa University of Naples, Italy E-mail: [email protected] Antonio Apicella University of Naples, Italy E-mail: [email protected] Taher M. Abu-Lebdeh North Carolina A and T State Univesity, United States E-mail: [email protected] Florian Ion Tiberiu Petrescu IFToMM, Romania E-mail: [email protected] Submission: 5/3/2019 Accept: 5/20/2019 ABSTRACT In other papers already presented on the structure and dimensions of elemental hydrogen, the elementary particle dynamics was taken into account in order to be able to determine the size of the hydrogen. This new work, one comes back with a new dynamic hypothesis designed to fundamentally change again the dynamic particle size due to the impulse influence of the particle. Until now it has been assumed that the impulse of an elementary particle is equal to the mass of the particle multiplied by its velocity, but in reality, the impulse definition is different, which is derived from the translational kinetic energy in a rapport of its velocity. This produces an additional condensation of matter in its elemental form. Keywords: Particle structure; Impulse; Condensed matter. [http://creativecommons.org/licenses/by/3.0/us/] Licensed under a Creative Commons Attribution 3.0 United States License 1749 INDEPENDENT JOURNAL OF MANAGEMENT & PRODUCTION (IJM&P) http://www.ijmp.jor.br v. -

Chemical Formulas the Elements
Chemical Formulas A chemical formula gives the numbers and types of atoms that are found in a substance. When the substance is a discrete molecule, then the chemical formula is also its molecular formula. Fe (iron) is a chemical formula Fe2O3 is a molecular formula The Elements The chemical formulas of most of the elements are simply their elemental symbol: Na (sodium) Fe (iron) He (helium) U (uranium) These chemical formulas are said to be monatomic—only an atom in chemical formula 1 The Elements There are seven elements that occur naturally as diatomic molecules—molecules that contain two atoms: H2 (hydrogen) N2 (nitrogen) O2 (oxygen) F2 (fluorine) Cl2 (chlorine) Br2 (bromine) I2 (iodine) The last four elements in this list are in the same family of the Periodic Table Binary Compounds A binary compound is one composed of only two different types of atoms. Rules for binary compound formulas 1. Element to left in Periodic Table comes first except for hydrogen: KCl PCl3 Al2S3 Fe3O4 2 Binary Compounds 2. Hydrogen comes last unless other element is from group 16 or 17: LiH, NH3, B2H4, CH4 3. If both elements are from the same group, the lower element comes first: SiC, BrF3 Other Compounds For compounds with three or more elements that are not ionic, if it contains carbon, this comes first followed by hydrogen. Other elements are then listed in alphabetical order: C2H6O C4H9BrO CH3Cl C8H10N4O2 3 Other Compounds However, the preceding rule is often ignored when writing organic formulas (molecules containing carbon, hydrogen, and maybe other elements) in order to give a better idea of how the atoms are connected: C2H6O is the molecular formula for ethanol, but nobody ever writes it this way—instead the formula is written C2H5OH to indicate one H atom is connected to the O atom. -

Syllabus of Biochemistry (Hons.)
Syllabus of Biochemistry (Hons.) for SEM-I & SEM-II under CBCS (to be effective from Academic Year: 2017-18) The University of Burdwan Burdwan, West Bengal 1 1. Introduction The syllabus for Biochemistry at undergraduate level using the Choice Based Credit system has been framed in compliance with model syllabus given by UGC. The main objective of framing this new syllabus is to give the students a holistic understanding of the subject giving substantial weight age to both the core content and techniques used in Biochemistry. The ultimate goal of the syllabus is that the students at the end are able to secure a job. Keeping in mind and in tune with the changing nature of the subject, adequate emphasis has been given on new techniques of mapping and understanding of the subject. The syllabus has also been framed in such a way that the basic skills of subject are taught to the students, and everyone might not need to go for higher studies and the scope of securing a job after graduation will increase. It is essential that Biochemistry students select their general electives courses from Chemistry, Physics, Mathematics and/or any branch of Life Sciences disciplines. While the syllabus is in compliance with UGC model curriculum, it is necessary that Biochemistry students should learn “Basic Microbiology” as one of the core courses rather than as elective while. Course on “Concept of Genetics” has been moved to electives. Also, it is recommended that two elective courses namely Nutritional Biochemistry and Advanced Biochemistry may be made compulsory. 2 Type of Courses Number of Courses Course type Description B. -

GANPAT UNIVERSITY FACULTY of SCIENCE TEACHING and EXAMINATION SCHEME Programme Bachelor of Science Branch/Spec
GANPAT UNIVERSITY FACULTY OF SCIENCE TEACHING AND EXAMINATION SCHEME Programme Bachelor of Science Branch/Spec. Microbiology Semester I Effective from Academic Year 2015- Effective for the batch Admitted in June 2015 16 Teaching scheme Examination scheme (Marks) Subject Subject Name Credit Hours (per week) Theory Practical Code Lecture(DT) Practical(Lab.) Lecture(DT) Practical(Lab.) CE SEE Total CE SEE Total L TU Total P TW Total L TU Total P TW Total UMBA101FOM FUNDAMENTALS 04 - 04 - - - 04 - 04 - - - 40 60 100 - - - OF MICROBIOLOGY UCHA101GCH GENERAL 04 - 04 - - - 04 - 04 - - - 40 60 100 - - - CHEMISTRY-I UPHA101GPH GENERAL 04 - 04 - - - 04 - 04 - - - 40 60 100 - - - PHYSICS-I UENA101ENG ENGLISH-I 02 - 02 - - - 02 - 02 - - - 40 60 100 - - - OPEN SUBJECT – 1 02 - 02 - - - 02 - 02 - - - 40 60 100 - - - UPMA101PRA PRACTICAL - - - 02 - 02 - - - 04 - 04 - - - - 50 50 MODULE-I UPCA101PRA PRACTICAL - - - 02 - 02 - - - 04 - 04 - - - - 50 50 MODULE-I UPPA101PRA PRACTICAL - - - 02 - 02 - - - 04 - 04 - - - - 50 50 MODULE-I Total 16 - 16 06 - 06 16 - 16 12 - 12 200 300 500 - 150 150 GANPAT UNIVERSITY FACULTY OF SCIENCE Programme Bachelor of Science Branch/Spec. Microbiology Semester I Version 1.0.1.0 Effective from Academic Year 2015-16 Effective for the batch Admitted in July 2015 Subject code UMBA 101 Subject Name FUNDAMENTALS OF MICROBIOLOGY FOM Teaching scheme Examination scheme (Marks) (Per week) Lecture(DT) Practical(Lab.) Total CE SEE Total L TU P TW Credit 04 -- -- -- 04 Theory 40 60 100 Hours 04 -- -- -- 04 Practical -- -- -- Pre-requisites: Students should have basic knowledge of Microorganisms and microscopy of 10+2 level. Learning Outcome: The course will help the student to understand basic fundamentals of Microbiology and history of Microbiology. -

HJ Lipkin the Weizmann Institute of Science Rehovot, Israel
WHO UNDERSTANDS THE ZWEIG-IIZUKA RULE? H.J. Lipkin The Weizmann Institute of Science Rehovot, Israel Abstract: The theoretical basis of the Zweig-Iizuka Ru le is discussed with the emphasis on puzzles and paradoxes which remain open problems . Detailed analysis of ZI-violating transitions arising from successive ZI-allowed transitions shows that ideal mixing and additional degeneracies in the particle spectrum are required to enable cancellation of these higher order violations. A quantitative estimate of the observed ZI-violating f' + 2rr decay be second order transitions via the intermediate KK state agrees with experiment when only on-shell contributions are included . This suggests that ZI-violations for old particles are determined completely by on-shell contributions and cannot be used as input to estimate ZI-violation for new particle decays where no on-shell intermediate states exist . 327 INTRODUCTION - SOME BASIC QUESTIONS 1 The Zweig-Iizuka rule has entered the folklore of particle physics without any clear theoretical understanding or justification. At the present time nobody really understands it, and anyone who claims to should not be believed. Investigating the ZI rule for the old particles raises many interesting questions which may lead to a better understanding of strong interactions as well as giving additional insight into the experimentally observed suppression of new particle decays attributed to the ZI rule. This talk follows an iconoclastic approach emphasizing embarrassing questions with no simple answers which might lead to fruitful lines of investigation. Both theoretical and experimental questions were presented in the talk. However, the experimental side is covered in a recent paper2 and is not duplicated here. -

Chemistry in One Dimension Pierre-Fran¸Coisloos,1, A) Caleb J
Chemistry in One Dimension Pierre-Fran¸coisLoos,1, a) Caleb J. Ball,1 and Peter M. W. Gill1, b) Research School of Chemistry, Australian National University, Canberra ACT 0200, Australia We report benchmark results for one-dimensional (1D) atomic and molecular sys- tems interacting via the Coulomb operator jxj−1. Using various wavefunction-type approaches, such as Hartree-Fock theory, second- and third-order Møller-Plesset per- turbation theory and explicitly correlated calculations, we study the ground state of atoms with up to ten electrons as well as small diatomic and triatomic molecules containing up to two electrons. A detailed analysis of the 1D helium-like ions is given and the expression of the high-density correlation energy is reported. We report the total energies, ionization energies, electron affinities and other interesting properties of the many-electron 1D atoms and, based on these results, we construct the 1D analog of Mendeleev's periodic table. We find that the 1D periodic table contains only two groups: the alkali metals and the noble gases. We also calculate the dissociation + 2+ 3+ curves of various 1D diatomics and study the chemical bond in H2 , HeH , He2 , + 2+ H2, HeH and He2 . We find that, unlike their 3D counterparts, 1D molecules are + primarily bound by one-electron bonds. Finally, we study the chemistry of H3 and we discuss the stability of the 1D polymer resulting from an infinite chain of hydrogen atoms. PACS numbers: 31.15.A-, 31.15.V-, 31.15.xt Keywords: one-dimensional atom; one-dimensional molecule; one-dimensional polymer; helium-like ions arXiv:1406.1343v1 [physics.chem-ph] 5 Jun 2014 a)Electronic mail: Corresponding author: [email protected] b)Electronic mail: [email protected] 1 I. -

Laser Cooling and Slowing of a Diatomic Molecule John F
Abstract Laser cooling and slowing of a diatomic molecule John F. Barry 2013 Laser cooling and trapping are central to modern atomic physics. It has been roughly three decades since laser cooling techniques produced ultracold atoms, leading to rapid advances in a vast array of fields and a number of Nobel prizes. Prior to the work presented in this thesis, laser cooling had not yet been extended to molecules because of their complex internal structure. However, this complex- ity makes molecules potentially useful for a wide range of applications. The first direct laser cooling of a molecule and further results we present here provide a new route to ultracold temperatures for molecules. In particular, these methods bridge the gap between ultracold temperatures and the approximately 1 kelvin temperatures attainable with directly cooled molecules (e.g. with cryogenic buffer gas cooling or decelerated supersonic beams). Using the carefully chosen molecule strontium monofluoride (SrF), decays to unwanted vibrational states are suppressed. Driving a transition with rotational quantum number R=1 to an excited state with R0=0 eliminates decays to unwanted ro- tational states. The dark ground-state Zeeman sublevels present in this specific scheme are remixed via a static magnetic field. Using three lasers for this scheme, a given molecule should undergo an average of approximately 100; 000 photon absorption/emission cycles before being lost via un- wanted decays. This number of cycles should be sufficient to load a magneto-optical trap (MOT) of molecules. In this thesis, we demonstrate transverse cooling of an SrF beam, in both Doppler and a Sisyphus-type cooling regimes.