An Autocrine Loop Involving Ret and Glial Cell–Derived Neurotrophic Factor Mediates Retinoic Acid–Induced Neuroblastoma Cell Differentiation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supp Material.Pdf
Legends for Supplemental Figures and Tables Figure S1. Expression of Tlx during retinogenesis. (A) Staged embryos were stained for β- galactosidase knocked into the Tlx locus to indicate Tlx expression. Tlx was expressed in the neural blast layer in the early phase of neural retina development (blue signal). (B) Expression of Tlx in neural retina was quantified using Q-PCR at multiple developmental stages. Figure S2. Expression of p27kip1 and cyclin D1 (Ccnd1) at various developmental stages in wild-type or Tlx-/- retinas. (A) Q-PCR analysis of p27kip1 mRNA expression. (B) Western blotting analysis of p27kip1 protein expression. (C) Q-PCR analysis of cyclin D1 mRNA expression. Figure S3. Q-PCR analysis of mRNA expression of Sf1 (A), Lrh1 (B), and Atn1 (C) in wild-type mouse retinas. RNAs from testis and liver were used as controls. Table S1. List of genes dysregulated both at E15.5 and P0 Tlx-/- retinas. Gene E15.5 P0 Cluste Gene Title Fold Fold r Name p-value p-value Change Change nuclear receptor subfamily 0, group B, Nr0b1 1.65 0.0024 2.99 0.0035 member 1 1 Pou4f3 1.91 0.0162 2.39 0.0031 POU domain, class 4, transcription factor 3 1 Tcfap2d 2.18 0.0000 2.37 0.0001 transcription factor AP-2, delta 1 Zic5 1.66 0.0002 2.02 0.0218 zinc finger protein of the cerebellum 5 1 Zfpm1 1.85 0.0030 1.88 0.0025 zinc finger protein, multitype 1 1 Pten 1.60 0.0155 1.82 0.0131 phospatase and tensin homolog 2 Itgb5 -1.85 0.0063 -1.85 0.0007 integrin beta 5 2 Gpr49 6.86 0.0001 15.16 0.0001 G protein-coupled receptor 49 3 Cmkor1 2.60 0.0007 2.72 0.0013 -

Gene Expression Profiles of Estrogen Receptor–Positive and Estrogen Receptor–Negative Breast Cancers Are Detectable in Histologically Normal Breast Epithelium
Published OnlineFirst November 8, 2010; DOI: 10.1158/1078-0432.CCR-10-1369 Clinical Cancer Human Cancer Biology Research Gene Expression Profiles of Estrogen Receptor–Positive and Estrogen Receptor–Negative Breast Cancers Are Detectable in Histologically Normal Breast Epithelium Kelly Graham1, Xijin Ge4, Antonio de las Morenas2, Anusri Tripathi3, and Carol L. Rosenberg1,2,3 Abstract Purpose: Previously, we found that gene expression in histologically normal breast epithelium (NlEpi) from women at high breast cancer risk can resemble gene expression in NlEpi from cancer-containing breasts. Therefore, we hypothesized that gene expression characteristic of a cancer subtype might be seen in NlEpi of breasts containing that subtype. Experimental Design: We examined gene expression in 46 cases of microdissected NlEpi from untreated women undergoing breast cancer surgery. From 30 age-matched cases [15 estrogen receptor (ER)þ,15ERÀ] we used Affymetryix U133A arrays. From 16 independent cases (9 ERþ,7ERÀ), we validated selected genes using quantitative real-time PCR (qPCR). We then compared gene expression between NlEpi and invasive breast cancer using four publicly available data sets. Results: We identified 198 genes that are differentially expressed between NlEpi from breasts with ERþ (NlEpiERþ) compared with ERÀ cancers (NlEpiERÀ). These include genes characteristic of ERþ and ERÀ cancers (e.g., ESR1, GATA3, and CX3CL1, FABP7). qPCR validated the microarray results in both the 30 original cases and the 16 independent cases. Gene expression in NlEpiERþ and NlEpiERÀ resembled gene expression in ERþ and ERÀ cancers, respectively: 25% to 53% of the genes or probes examined in four external data sets overlapped between NlEpi and the corresponding cancer subtype. -

Supplementary Table 1: Genes Affected by Anoikis. A, Ratio of Signal
Supplementary Table 1: Genes affected by anoikis. a, ratio of signal intensity of nonanchored cells anchorage dependent cells (CasKoSrc) over anchored cells; b, induced by Src transformation of Cx43KO cells; c, decreased by Src transformation of Cx43Ko cells; *, induced by normalization of Src transformed cells by neighboring nontransformed cells. Gene Symbol Probe Set Fold Changea Gene Name increased Selenbp1 1450699_at 23.22 selenium binding protein 1 Dscr1l1 1450243_a_at 10.77 Down syndrome critical region gene 1-like 1 Dscr1l1 1421425_a_at 4.29 Down syndrome critical region gene 1-like 1 Ttyh1 1426617_a_at 6.70 tweety homolog 1 (Drosophila) 5730521E12Rik 1419065_at 6.16 RIKEN cDNA 5730521E12 gene c 6330406I15Rik 1452244_at 5.87 RIKEN cDNA 6330406I15 gene AF067063 1425160_at 5.73 clone L2 uniform group of 2-cell-stage gene family mRNA Morc 1419418_a_at 5.55 microrchidia c Gpr56 1421118_a_at 5.43 G protein-coupled receptor 56 Pax6 1452526_a_at 5.06 paired box gene 6 Tgfbi 1415871_at 3.73 transforming growth factor beta induced Adarb1 1434932_at 3.70 adenosine deaminase RNA-specific B1 Ddx3y 1452077_at 3.30 DEAD (Asp-Glu-Ala-Asp) box polypeptide 3 Y-linked b Ampd3 1422573_at 3.20 AMP deaminase 3 Gli2 1459211_at 3.07 GLI-Kruppel family member GLI2 Selenbp2 1417580_s_at 2.96 selenium binding protein 2 Adamts1 1450716_at 2.80 a disintegrin-like and metalloprotease with thrombospondin type 1 motif 1 Dusp15 1426189_at 2.70 dual specificity phosphatase-like 15 Dpep3 1429035_at 2.60 dipeptidase 3 Sepp1 1452141_a_at 2.57 selenoprotein P plasma -

Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients with Stable Coronary Heart Disease
Supplementary Online Content Ganz P, Heidecker B, Hveem K, et al. Development and validation of a protein-based risk score for cardiovascular outcomes among patients with stable coronary heart disease. JAMA. doi: 10.1001/jama.2016.5951 eTable 1. List of 1130 Proteins Measured by Somalogic’s Modified Aptamer-Based Proteomic Assay eTable 2. Coefficients for Weibull Recalibration Model Applied to 9-Protein Model eFigure 1. Median Protein Levels in Derivation and Validation Cohort eTable 3. Coefficients for the Recalibration Model Applied to Refit Framingham eFigure 2. Calibration Plots for the Refit Framingham Model eTable 4. List of 200 Proteins Associated With the Risk of MI, Stroke, Heart Failure, and Death eFigure 3. Hazard Ratios of Lasso Selected Proteins for Primary End Point of MI, Stroke, Heart Failure, and Death eFigure 4. 9-Protein Prognostic Model Hazard Ratios Adjusted for Framingham Variables eFigure 5. 9-Protein Risk Scores by Event Type This supplementary material has been provided by the authors to give readers additional information about their work. Downloaded From: https://jamanetwork.com/ on 10/02/2021 Supplemental Material Table of Contents 1 Study Design and Data Processing ......................................................................................................... 3 2 Table of 1130 Proteins Measured .......................................................................................................... 4 3 Variable Selection and Statistical Modeling ........................................................................................ -

Ssecks/Gravin/AKAP12 Attenuates Expression of Proliferative And
BMC Cancer BioMed Central Research article Open Access SSeCKS/Gravin/AKAP12 attenuates expression of proliferative and angiogenic genes during suppression of v-Src-induced oncogenesis Yongzhong Liu1, Lingqiu Gao2 and Irwin H Gelman*2 Address: 1Mucosal Immunology Unit, National Institutes of Health, Bethesda, MD 20892, USA and 2Department of Cancer Genetics, Roswell Park Cancer Institute, Buffalo, NY 14263, USA Email: Yongzhong Liu - [email protected]; Lingqiu Gao - [email protected]; Irwin H Gelman* - [email protected] * Corresponding author Published: 25 April 2006 Received: 24 January 2006 Accepted: 25 April 2006 BMC Cancer2006, 6:105 doi:10.1186/1471-2407-6-105 This article is available from: http://www.biomedcentral.com/1471-2407/6/105 © 2006Liu et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: SSeCKS is a major protein kinase C substrate with kinase scaffolding and metastasis- suppressor activity whose expression is severely downregulated in Src- and Ras-transformed fibroblast and epithelial cells and in human prostate, breast, and gastric cancers. We previously used NIH3T3 cells with tetracycline-regulated SSeCKS expression plus a temperature-sensitive v-Src allele to show that SSeCKS re-expression inhibited parameters of v-Src-induced oncogenic growth without attenuating in vivo Src kinase activity. Methods: We use cDNA microarrays and semi-quantitative RT-PCR analysis to identify changes in gene expression correlating with i) SSeCKS expression in the absence of v-Src activity, ii) activation of v-Src activity alone, and iii) SSeCKS re-expression in the presence of active v-Src. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

A Role for Glial Cell–Derived Neurotrophic Factor–Induced Expression by Inflammatory Cytokines and RET/GFRA1 Receptor Up-Regulation in Breast Cancer
Research Article A Role for Glial Cell–Derived Neurotrophic Factor–Induced Expression by Inflammatory Cytokines and RET/GFRA1 Receptor Up-regulation in Breast Cancer Selma Esseghir,1 S. Katrina Todd,1 Toby Hunt,2 Richard Poulsom,2 Ivan Plaza-Menacho,1 Jorge S. Reis-Filho,1 and Clare M. Isacke1 1Breakthrough Breast Cancer Research Centre, The Institute of Cancer Research and 2In Situ Hybridisation Service and Histopathology Unit, Cancer Research UK-London Research Institute, London, United Kingdom Abstract system homeostasis and the inflammatory response (3, 4) and in By screening a tissue microarray of invasive breast tumors, we tumor progression (5). The neurotrophic factor glial cell–derived have shown that the receptor tyrosine kinase RET (REar- neurotrophic factor (GDNF) was first identified for its trophic ranged during Transfection) and its coreceptor GFRA1(GDNF activity on midbrain dopaminergic neurons; however, GDNF has receptor family A-1) are overexpressed in a subset of estrogen subsequently been shown to have broader effects in regulating receptor–positive tumors. Germ line–activating oncogenic growth, survival, and migration of neurons in the brain, spinal cord, mutations in RET allow this receptor to signal independently and periphery as well as having an essential role in the growth and A branching of the ureteric buds of the developing kidney (6, 7). Mice of GFR 1and its ligand glial cell–derived neurotrophic factor Gdnf (GDNF) to promote a spectrum of endocrine neoplasias. with a homozygous deletion of die shortly after birth due to However, it is not known whether tumor progression can also severe defects in renal differentiation and the absence of an enteric nervous system (8, 9). -

Taqman® Human Protein Kinase Array
TaqMan® Gene Signature Arrays TaqMan® Human Protein Kinase Array This array is part of a collection of TaqMan® Gene Signature these kinases are from receptor protein-tyrosine kinase (RPTK) Arrays that enable analysis of hundreds of TaqMan® Gene families: EGFR, InsulinR, PDGFR, VEGFR, FGFR, CCK, NGFR, Expression Assays on a micro fluidic card with minimal effort. HGFR, EPHR, AXL, TIE, RYK, DDR, RET, ROS, LTK, ROR and MUSK. The remaining 15 kinases are Ser/Thr kinases from the Protein kinases are one of the largest families of genes in kinase families: CAMKL, IRAK, Lmr, RIPK and STKR. eukaryotes. They belong to one superfamily containing a eukaryotic protein kinase catalytic domain. The ability of kinases We have also selected assays for 26 non-kinase genes in the to reversibly phosphorylate and regulate protein function Human Protein Kinase Array. These genes are involved in signal has been a subject of intense investigation. Kinases are transduction and mediate protein-protein interaction, transcrip- responsible for most of the signal transduction in eukaryotic tional regulation, neural development and cell adhesion. cells, affecting cellular processes including metabolism, References: angiogenesis, hemopoiesis, apoptosis, transcription and Manning, G., Whyte, D.B., Martinez, R., Hunter, T., and differentiation. Protein kinases are also involved in functioning Sudarsanam, S. 2002. The Protein Kinase Complement of the of the nervous and immune systems, in physiologic responses Human Genome. Science 298:1912–34. and in development. Imbalances in signal transduction due to accumulation of mutations or genetic alterations have Blume-Jensen, P. and Hunter, T. 2001. Oncogenic kinase been shown to result in malignant transformation. -

The Neuroprotective Role of the GM1 Oligosaccharide, Ii3neu5ac-Gg4, In
Molecular Neurobiology (2019) 56:6673–6702 https://doi.org/10.1007/s12035-019-1556-8 The Neuroprotective Role of the GM1 Oligosaccharide, 3 II Neu5Ac-Gg4, in Neuroblastoma Cells Elena Chiricozzi1 & Margherita Maggioni1 & Erika di Biase1 & Giulia Lunghi1 & Maria Fazzari1 & Nicoletta Loberto 1 & Maffioli Elisa2 & Francesca Grassi Scalvini2 & Gabriella Tedeschi 2,3 & Sandro Sonnino1 Received: 10 January 2019 /Accepted: 13 March 2019 /Published online: 26 March 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract 3 Recently, we demonstrated that the GM1 oligosaccharide, II Neu5Ac-Gg4 (OligoGM1), administered to cultured murine Neuro2a neuroblastoma cells interacts with the NGF receptor TrkA, leading to the activation of the ERK1/2 downstream pathway and to cell differentiation. To understand how the activation of the TrkA pathway is able to trigger key biochemical signaling, we performed a proteomic analysis on Neuro2a cells treated with 50 μM OligoGM1 for 24 h. Over 3000 proteins were identified. Among these, 324 proteins were exclusively expressed in OligoGM1-treated cells. Interestingly, several proteins expressed only in OligoGM1-treated cells are involved in biochemical mechanisms with a neuroprotective potential, reflecting the GM1 neuroprotective effect. In addition, we found that the exogenous administration of OligoGM1 reduced the cellular oxidative stress in Neuro2a cells and conferred protection against MPTP neurotoxicity. These results confirm and reinforce the idea that the molecular mechanisms underlying the GM1 neurotrophic and neuroprotective effects depend on its oligosaccharide chain, suggesting the activation of a positive signaling starting at plasma membrane level. Keywords GM1 ganglioside . GM1 oligosaccharide chain . TrkA neurotrophin receptor . Plasma membrane signaling . Neuroprotection . -
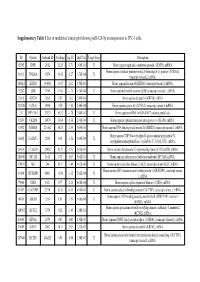
Supplementary Table 1 List of Modulated Transcripts Following Mir-129-5P Overexpression in TPC-1 Cells
Supplementary Table 1 List of modulated transcripts following miR-129-5p overexpression in TPC-1 cells. ID Symbol Genbank ID AveExpr log_FC adj.P.Val Target Scan Description 92585 EMP1 2012 12.15 -3.71 1.06E-04 X Homo sapiens epithelial membrane protein 1 (EMP1), mRNA. Homo sapiens vacuolar protein sorting 26 homolog A (S. pombe) (VPS26A), 10111 VPS26A 9559 11.92 -2.27 1.70E-04 X transcript variant 2, mRNA. 186912 KRT80 144501 11.97 2.62 1.70E-04 Homo sapiens keratin 80 (KRT80), transcript variant 2, mRNA. 92202 LBR 3930 11.86 -1.75 1.70E-04 X Homo sapiens lamin B receptor (LBR), transcript variant 1, mRNA. 33816 KRT34 3885 7.87 1.62 2.48E-04 Homo sapiens keratin 34 (KRT34), mRNA. 121520 CCNA1 8900 9.50 -1.80 2.48E-04 Homo sapiens cyclin A1 (CCNA1), transcript variant 4, mRNA. 211 DPY19L1 23333 10.32 -1.78 2.48E-04 X Homo sapiens mRNA for KIAA0877 protein, partial cds. 15854 CKAP4 10970 13.04 -1.36 2.66E-04 X Homo sapiens cytoskeleton-associated protein 4 (CKAP4), mRNA. 63392 RBM24 221662 10.83 2.39 3.09E-04 Homo sapiens RNA binding motif protein 24 (RBM24), transcript variant 2, mRNA. Homo sapiens UDP-N-acetyl-alpha-D-galactosamine:polypeptide N- 36665 GALNT1 2589 9.61 -1.52 3.09E-04 X acetylgalactosaminyltransferase 1 (GalNAc-T1) (GALNT1), mRNA. 18614 C12orf24 29902 10.57 1.76 5.02E-04 Homo sapiens chromosome 12 open reading frame 24 (C12orf24), mRNA. 126848 MCAM 4162 8.72 2.05 5.02E-04 X Homo sapiens melanoma cell adhesion molecule (MCAM), mRNA. -

A Dual-Strategy Expression Screen for Candidate Connectivity Labels in the Developing Thalamus
bioRxiv preprint doi: https://doi.org/10.1101/079327; this version posted October 5, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Connectivity labels in developing thalamus A dual-strategy expression screen for candidate connectivity labels in the developing thalamus Olivia Bibollet-Bahena1, Tatsuya Okafuji1, Karsten Hokamp1 and Kevin J. Mitchell1,2 1Smurfit Institute of Genetics 2Institute of Neuroscience Trinity College Dublin Dublin 2, Ireland Address for correspondence: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/079327; this version posted October 5, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Connectivity labels in developing thalamus Abstract The thalamus or “inner chamber” of the brain is divided into ~30 discrete nuclei, with highly specific patterns of afferent and efferent connectivity. To identify genes that may direct these patterns of connectivity, we used two strategies. First, we used a bioinformatics pipeline to survey the predicted proteomes of nematode, fruitfly, mouse and human for extracellular proteins containing any of a list of motifs found in known guidance or connectivity molecules. Second, we performed clustering analyses on the Allen Developing Mouse Brain Atlas data to identify genes encoding surface proteins expressed with temporal profiles similar to known guidance or connectivity molecules. -

A Pharmacological Interactome Platform for Discovery of Pain Mechanisms and Targets
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.14.041715; this version posted April 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. A pharmacological interactome platform for discovery of pain mechanisms and targets Short title: A pharmacological interactome for pain target identification Andi Wangzhou1, Candler Paige1, Sanjay V Neerukonda1, Gregory Dussor1, Pradipta R Ray1,#, Theodore J Price1,#,* 1The University of Texas at Dallas, School of Behavioral and Brain Sciences and Center for Advanced Pain Studies, 800 W Campbell Rd. Richardson, TX, 75080, USA # corresponding authors [email protected] and [email protected] * lead contact Funding: NIH grants NS113457 (CP), NS065926 (TJP) and NS102161 (TJP) The authors declare no conflicts of interest. Author Contributions: Conceived of the Project: GD, PRR and TJP Performed Experiments: AW, CP, SVN and PRR Supervised Experiments: GD, PRR and TJP Analyzed Data: AW, CP and PRR Drew Figures: AW, and CP Wrote and Edited Manuscript: AW, CP, GD, PRR and TJP All authors approved the final version of the manuscript. Acknowledgements: The authors would like to thank Drs. James Hockley and Ewan St. John Smith for help with the colonic single neuron sequencing data, Dr. Brian Gulbransen and lab for help with the enteric glia TRAP data and Dr. Zhenyu Xuan for clarifying TCGA metadata formats. We thank all the authors of the papers from which we used their sequencing data for their exemplary transparency in sharing the details of their work with us.