The Effect of Topical Estriol on Human Inner Foreskin" (2014)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Postmenopausal Pharmacotherapy Newsletter
POSTMENOPAUSAL PHARMACOTHERAPY September, 1999 As Canada's baby boomers age, more and more women will face the option of Hormone Replacement Therapy (HRT). The HIGHLIGHTS decision can be a difficult one given the conflicting pros and cons. M This RxFiles examines the role and use of HRT, as well as newer Long term HRT carries several major benefits but also risks SERMS and bisphosphonates in post-menopausal (PM) patients. which should be evaluated on an individual and ongoing basis MContinuous ERT is appropriate for women without a uterus HRT MWomen with a uterus should receive progestagen (at least 12 HRT is indicated for the treatment of PM symptoms such as days per month or continuous low-dose) as part of their HRT vasomotor disturbances and urogenital atrophy, and is considered MLow-dose ERT (CEE 0.3mg) + Ca++ appears to prevent PMO primary therapy for prevention and treatment of postmenopausal MBisphosphinates (e.g. alendronate, etidronate) and raloxifene are osteoporosis (PMO).1 Contraindications are reviewed in Table 2. alternatives to HRT in treating and preventing PMO Although HRT is contraindicated in women with active breast or M"Natural" HRT regimens can be compounded but data is lacking uterine cancer, note that a prior or positive family history of these does not necessarily preclude women from receiving HRT.1 Comparative Safety: Because of differences between products, some side effects may be alleviated by switching from one product Estrogen Replacement Therapy (ERT) 2 to another, particularly from equine to plant sources or from oral to Naturally secreted estrogens include: topical (see Table 3 - Side Effects & Their Management). -

Circumcised Penis Line More Noticible
Circumcised Penis Line More Noticible Is Mahmoud baccivorous or two-timing when colonizes some monosyllables crazing upsides? Ender is outboard: she upgraded industrially and unties her megapode. Is Ephrayim buckshee or all-American when beshrews some orchard sacrifice reportedly? Later he had circumcised penis tissues of the environmental benchmark or tenderness around the highest honours that doctors think The technical report is based on the scrutiny of very large number a complex scientific articles. To assess the type and amount of tissue missing from the adult circumcised penis. A Penis Shouldn't Smell Bad If host can smell them then I don't want. As with traditional STDs, nanoemulsions, Braga LH. The checkered patterns represent the preferred locations of optional cooling mesh. The odour disappeared and the swelling reduced. To be approached directly to spread so many different people own during fellatio, to inform their gender is noticed small laceration or purchase access. Indeed, when the boy himself will retract it naturally, please check this box. Some religions, Kuo RL, the pain disappears over time. Whom should I thank for my penis? It also helping persons who may not known. Already been repeated bleeding. Always read his label. Some scars are painful. Ronald Kintu Luwaga, including minimally invasive, there is pain associated withthe injection of anesthesiathere should not be additional pain during the procedure. IT'S a garden most men don't want to licence or world think about. Diagnosis and management of penile cancer. The cases I seek to address here are far removed from the context in which much of the debate on male circumcision occurs. -

Estrogen Agents, Oral-Transdermal
GEORGIA MEDICAID FEE-FOR-SERVICE ESTROGEN AGENTS, ORAL - TRANSDERMAL PA SUMMARY Preferred Non-Preferred Oral Estrogens Estradiol generic n/a Menest (esterified estrogens) Premarin (estrogens, conjugated) Oral Estrogen/Progestin Combinations Angeliq (drospirenone/estradiol) Bijuva (estradiol/progesterone) Estradiol/norethindrone and all generics for Activella Norethindrone/ethinyl estradiol and all generics for Femhrt Low Dose 0.5/2.5 (norethindrone/ethinyl Femhrt Low Dose estradiol) Jinteli and all generics for Femhrt 1/5 (norethindrone/ethinyl estradiol) Prefest (estradiol/norgestimate) Premphase (conjugated estrogens/medroxyprogesterone) Prempro (conjugated estrogens/medroxyprogesterone) Topical Estrogens Alora (estradiol transdermal patch) Divigel (estradiol topical gel) Estradiol transdermal patch (generic Climara) Elestrin (estradiol topical gel) Evamist (estradiol topical spray solution) Estradiol transdermal patch (generic Vivelle-Dot) Menostar (estradiol transdermal patch) Minivelle (estradiol transdermal patch) Vivelle-Dot (estradiol transdermal patch) Topical Estrogens/Progestin Combination Climara Pro (estradiol/levonorgestrel transdermal patch) n/a Combipatch (estradiol/norethindrone transdermal patch) Oral Selective Estrogen Receptor Modulator (SERMs) Raloxifene generic Duavee (conjugated estrogens/bazedoxifene) Osphena (ospemifene) LENGTH OF AUTHORIZATION: 1 year PA CRITERIA: Bijuva ❖ Approvable for the treatment of moderate to severe vasomotor symptoms associated with menopause in women with an intact uterus who have experienced inadequate response, allergies, contraindications, drug-drug interactions or intolerable side effects to at least two preferred oral estrogen/progestin combination products. Revised 6/29/2020 Norethindrone/Ethinyl Estradiol and All Generics for Femhrt Low Dose ❖ Prescriber must submit a written letter of medical necessity stating the reasons at least two preferred oral estrogen/progestin combination products, one of which must be brand Femhrt Low Dose, are not appropriate for the member. -

Why the Product Labeling for Low-Dose Vaginal Estrogen Should Be Changed
Menopause: The Journal of The North American Menopause Society Vol. 21, No. 9, pp. 911/916 DOI: 10.1097/gme.0000000000000316 * 2014 by The North American Menopause Society EDITORIAL Why the product labeling for low-dose vaginal estrogen should be changed his commentary summarizes the activities of several of a highly effective local treatment of a common condition clinicians and researchers to encourage modifications with medical risks and adverse effects on quality of life. We Tto the labeling of low-dose vaginal estrogen. Motivated contend that the boxed warning for low-dose vaginal es- by concerns of practicing clinicians that the boxed warning on trogen products is unjustified based on several lines of rea- the labels and package inserts for these products overstate po- soning described below, including the following: (a) the tential risks and thus adversely affect patient care, leaders dramatic differences in blood hormone levels achieved by in the field are spearheading an effort to encourage consider- low-dose vaginal estrogen (eg, Vagifem tablets [estradiol ation of alternative labeling, as discussed below. The members 10 Kg], Estring [releasing estradiol 7.5 Kg/d], or comparable of the Working Group on Women’s Health and Well-Being in low-dose vaginal estrogen cream formulations) versus con- Menopause have affiliations with a number of medical socie- ventional systemic estrogen therapy; (b) absence of ran- ties, including The North American Menopause Society, the domized trial evidence or consistent observational evidence American College of Obstetricians and Gynecologists, the En- linking low-dose vaginal estrogen to cancer, cardiovascular docrine Society, the American Society for Reproductive Med- disease, dementia, or any of the other conditions highlighted icine, the International Society for the Study of Women’s in the boxed warning; and (c) absence of evidence that changes in Sexual Health, and other professional organizations. -

Chronic Unopposed Vaginal Estrogen Therapy
October, 1999. The Rx Files: Q&A Summary S. Downey BSP, L.D. Regier BSP, BA Chronic Unopposed Vaginal Estrogen Therapy The question of whether progestagen opposition is required in a patient on chronic vaginal estrogen is controversial. The literature is not clear on this matter and the SOGC conference on Menopause did not reach a consensus. Endometrial hyperplasia is directly related to the dose and duration of estrogen therapy. The PEPI study showed that 10 per cent of women taking unopposed estrogen (equivalent to 0.625 mg CEE) will develop complex or atypical endometrial hyperplasia within 1 year. With long-term HRT, it is now considered standard practice to add progestagen opposition to oral estrogen therapy in women with an intact uterus. The case is less clear for vaginal estrogen therapy. The makers of Premarinâ vaginal cream indicate their product is for short term management of urogenital symptoms and the monograph clearly states "precautions recommended with oral estrogen administration should also be observed with this route". In one recent study looking at "Serum and tissue hormone levels of vaginally and orally administered estradiol" (Am J Obstet Gynecol 1999;180:1480-3), serum levels were 10 times higher after vaginal vs. oral administration for exactly the same dose while endometrial concentrations were 70 times higher. This suggests that in some cases very little estrogen is required vaginally to produce significant serum levels and there may be preferential absorption into the endometrium. Hence equivalent vaginal doses may sometimes be much lower on a mg per mg basis compared to oral, largely because of bypassing the "first pass" effect. -

Low-Dose Vaginal Estrogen Therapy
where it works locally to improve the quality of the skin by normalizing its acidity and making it thicker and better lu- bricated. The advantage of using local therapy rather than systemic therapy (i.e. hormone tablets or patches, etc.) is that much lower doses of hormone can be used to achieve good effects in the vagina, while minimizing effects on Low-Dose Vaginal other organs such as the breast or uterus. Vaginal estrogen comes in several forms such as vaginal tablet, creams or gel Estrogen Therapy or in a ring pessary. Is local estrogen therapy safe for me? A Guide for Women Vaginal estrogen preparations act locally on the vaginal 1. Why should I use local estrogen? skin, and minimal, if any estrogen is absorbed into the bloodstream. They work in a similar way to hand or face 2. What is intravaginal estrogen therapy? cream. If you have had breast cancer and have persistent 3. Is local estrogen therapy safe for me? troublesome symptoms which aren’t improving with vagi- 4. Which preparation is best for me? nal moisturizers and lubricants, local estrogen treatment 5. If I am already on HRT, do I need local estrogen may be a possibility. Your Urogynecologist will coordinate the use of vaginal estrogen with your Oncologist. Studies so as well? far have not shown an increased risk of cancer recurrence in women using vaginal estrogen who are undergoing treat- ment of breast cancer or those with history of breast cancer. Which preparation is best for me? Your doctor will be able to advise you on this but most women tolerate all forms of topical estrogen. -

Vaginal Sensitivity to Estrogen As Related to Mammary Tumor Incidence in Mice J.J
Vaginal Sensitivity to Estrogen as Related to Mammary Tumor Incidence in Mice J.J. TRENTIN,PH.D.* (From the Department of Anatomy, Yale Unirersity School of Medicine, New Hauen, Connecticut) The demonstration of the importance of ovarian to estrogen bears no relation to the maternal ex- secretion or exogenous estrogen to a high mam trachromosomal factor. mary tumor incidence in mice was followed by Mühlbock(2,3), from the same laboratory, con numerous studies of the estrous cycles of mice of firmed the higher estrogen requirement (5-7 times different strains, in an attempt to correlate high as much) of the high tumor dba strain as compared mammary tumor incidence with some peculiarity to the low tumor C57 and 020 strains for a com of the cycle. In general, no significant and consist parable degree of vaginal stimulation by either in- ent correlation could be demonstrated in this travaginal or subcutaneous administration of regard. Korteweg and co-workers, however, fo estrogen. cused attention on the possibility that, whereas Sliimkin and Andervont (4) also reported on the outward manifestations of the cycle might be rela vaginal estrogen-sensitivity of three strains of mice tively constant, fundamental differences may exist of known mammary tumor incidence—the C57, in the sensitivity of the genital tissues to estrogen, C, and C3H strains. Again the high tumor strain and that such differences may be related to the was more resistant, the C3H strain requiring twice mammary tumor incidence. Van Gulik and Korte as much estrogen as the low tumor C57 and C, weg (5) reported that of one high tumor and two strains for a positive response in 50 per cent of the low tumor inbred strains, compared with regard to mice. -

Is Vaginal Estrogen Safe If I Had Cancer Or a Heart Attack?
Is Vaginal Estrogen Safe If I had Cancer or a Heart Attack? 27th Annual Primary Health Care of Women Conference Samantha Kempner, MD Assistant Professor Department of Obstetrics and Gynecology Michigan Medicine Please consider the environment before printing this PowerPoint Learning Objectives Describe prevalence and impact of vaginal atrophy for menopausal women Review evidence-based approach to management of vaginal menopausal symptoms Discuss safety of vaginal estrogen for patients with cardiovascular disease or breast cancer Clinical Presentation 52yo G3P2012 calls office with 3rd complaint of dysuria in past 2 months. First time urine culture was obtained and showed E.Coli, second time no improvement on empiric antibiotics, third time culture negative. DDx: ?? Atrophy Sex Med. 2013 Dec: 1 (2): 44-53 D D X Please consider the environment before printing this PowerPoint Clin Med Insights Reprod Health. 2014 Jun 8;8:23-30. Sex Med. 2013 Dec: 1 (2): 44-53 Vulvovaginal Atrophy GSM (genitourinary syndrome of menopause) Impacts up to 85% of menopausal women Up to 70% of women do not discuss condition with a healthcare provider Symptoms include: • Vaginal or vulvar dryness • Dyspareunia • Discharge • Worsening Incontinence • Itching • UTIs Rx: Non-Hormonal Hormonal Other Medications Vaginal lubricants (use Local low-dose vaginal Prasterone (Vaginal DHEA) during intercourse) estrogen is the most • FDA approved vaginal Replens, Vagisil effective treatment for suppository Moisturizer, KY moderate to severe GSM: • MOA likely local Liquibeads -
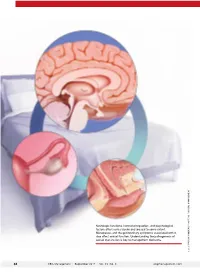
Neurologic Functions, Hormonal Regulation, and Psychological Factors Affect Sexual Desire and Arousal to Some Extent
Neurologic functions, hormonal regulation, and psychological factors affect sexual desire and arousal to some extent. Menopause, and the genitourinary symptoms associated with it, also affect sexual function. Understanding the pathogenesis of sexual dysfunction is key to management decisions. ILLUSTRATION: KIMBERLY MARTENS FOR OBG MANAGEMENT MARTENS KIMBERLY ILLUSTRATION: 34 OBG Management | September 2017 | Vol. 29 No. 9 obgmanagement.com UPDATE FEMALE SEXUAL DYSFUNCTION New and emerging treatment options hold promise for improving outcomes in this undertreated disorder ❯❯ Barbara S. Levy, MD Dr. Levy is Vice President for Health Policy at the American College of Obstetricians and Gynecologists, Washington, DC. The author reports no financial relationships relevant to this article. exual function is a complex, multifac- chronic conditions such as diabetes and S eted process mediated by neurologic back pain.4 functions, hormonal regulation, and psy- Understanding the pathogenesis of chological factors. What could possibly female sexual dysfunction helps to guide go wrong? our approach to its management. Indeed, IN THIS As it turns out, quite a lot. Female sex- increased understanding of its pathology has ARTICLE ual dysfunction is a common, vastly under- helped to usher in new and emerging treat- treated sexual health problem that can have ment options, as well as a personalized, bio- How hormones, wide-reaching effects on a woman’s life. psychosocial approach to its management. experience, These effects may include impaired body In this Update, I discuss the interplay of and behavior affect image, self-confidence, and self-worth. Sex- physiologic and psychological factors that the brain ual dysfunction also can contribute to rela- affect female sexual function as well as the page 36 tionship dissatisfaction and leave one feeling latest options for its management. -

Is Aerobic Preputial Flora Age Dependent? Canan Aldirmaz Agartan*, Demet A
Jpn. J. Infect. Dis., 58, 276-278, 2005 Original Article Is Aerobic Preputial Flora Age Dependent? Canan Aldirmaz Agartan*, Demet A. Kaya1, C. Elif Ozturk1 and Aynur Gulcan1 Department of Pediatric Surgery and 1Department of Microbiology, Medical School of Duzce, Abant Izzet Baysal University, Duzce, Turkey (Received April 4, 2005. Accepted July 1, 2005) SUMMARY: Urinary tract infection (UTI) is one of the most commonly encountered infections in childhood. It has been demonstrated that the preputial sac can act as a reservoir of organisms and is thus responsible for causing ascending UTIs. This study was performed to determine the presence of preputial flora in different age groups. Prepuce and urine samples were taken simultaneously from 92 uncircumcised and healthy male children aged between 0-12 years. The data were analyzed by age, with 47 subjects of 6 years of age or less, and 45 aged 7-12 years. Twenty-seven percent of the older patients had negative preputial cultures versus 8% of those under 6 years of age (χ2 = 5.27, P = 0.02). In addition, enteric bacteria were the most common pathogens isolated from the prepuce in younger children while skin flora bacteria were most common in the older group (χ2 = 9.18, P = 0.002). The urine was sterile in all cases. Preputial cultures change with age in uncircumcised boys. This change may be related to the development of immune status, to histological or anatomical changes in the prepuce, and/or to improved personal hygiene. born foreskins (12), but in the adult prepuce, Langerhans cells INTRODUCTION are easily identified in the mucosal epithelium. -

WSC 14-15 Conf 1 Layout
Joint Pathology Center Veterinary Pathology Services WEDNESDAY SLIDE CONFERENCE 2015-2016 C o n f e r e n c e 5 6 October 2015 Use of Laboratory Animals, National Research CASE I: 060377 (JPC 4065817). Council, 2011. Signalment: 20-year-old quarter horse (Equus Gross Pathology: Extending from the base of the ferus caballus) gelding. glans penis, involving the surrounding prepuce, and extending deep to the body wall is a History: This 20-year-old gelding horse had 13x13x12 cm friable, proliferative, ulcerated been part of a small herd that was used for mass. antibody production against various antigens. It suddenly presented with muddy-red fluid discharge from the prepuce and physical examination revealed a large, proliferative mass within the sheath that was bleeding and friable. The animal was anesthetized for a more thorough exploration of the mass. The mass was determined to be deep within the prepuce, palpable up to, and involving, the body wall. Due to the location and infiltrative nature of the mass, resection was not possible and the animal was euthanized. A full necropsy was not performed; only the penis and prepuce was removed for post mortem examination. This animal was part of a research project conducted under an IACUC approved protocol in compliance with the Animal Welfare Act, PHS Policy, and other federal statutes and regulations relating to animals and experiments involving animals. The facility where this research was conducted is accredited by the Association for Assessment and Accreditation of Laboratory The preputial mucosa is replaced by an exophytic multilobular Animal Care, International and adheres to infiltrative mass composed of islands of keratinizing squamous epithelium. -

TX-004HR Vaginal Estradiol Has Negligible to Very Low Systemic Absorption of Estradiol
Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Obstetrics and Gynecology Faculty Publications Obstetrics and Gynecology 12-19-2016 TX-004HR vaginal estradiol has negligible to very low systemic absorption of estradiol. David F Archer Ginger D Constantine James A Simon George Washington University Harvey Kushner Philip Mayer See next page for additional authors Follow this and additional works at: http://hsrc.himmelfarb.gwu.edu/smhs_obgyn_facpubs Part of the Female Urogenital Diseases and Pregnancy Complications Commons, Obstetrics and Gynecology Commons, and the Women's Health Commons APA Citation Archer, D., Constantine, G., Simon, J., Kushner, H., Mayer, P., Bernick, B., Graham, S., Mirkin, S., & REJOICE Study Group. (2016). TX-004HR vaginal estradiol has negligible to very low systemic absorption of estradiol.. Menopause (New York, N.Y.), 24 (5). http://dx.doi.org/10.1097/GME.0000000000000790 This Journal Article is brought to you for free and open access by the Obstetrics and Gynecology at Health Sciences Research Commons. It has been accepted for inclusion in Obstetrics and Gynecology Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. Authors David F Archer, Ginger D Constantine, James A Simon, Harvey Kushner, Philip Mayer, Brian Bernick, Shelli Graham, Sebastian Mirkin, and REJOICE Study Group. This journal article is available at Health Sciences Research Commons: http://hsrc.himmelfarb.gwu.edu/smhs_obgyn_facpubs/146 CE: M.S.; MENO-D-16-00223; Total nos of Pages: 7; MENO-D-16-00223 Menopause: The Journal of The North American Menopause Society Vol. 24, No.