Isbister Et Al 2014.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

False Black Widows and Other Household Spiders
False Black Widows and Other Household Spiders Spiders can quite unnecessarily evoke all kinds of dread and fear. The Press does not help by publishing inaccurate and often alarmist stories about them. Spiders are in fact one of our very important beneficial creatures. Spiders in the UK devour a weight of insect 'pests' equivalent to that of the nation's human population! During the mid-late summer, many spiders mature and as a result become more obvious as they have then grown to their full size. One of these species is Steatoda nobilis. It came from the Canary and Madeiran Islands into Devon over a 100 years ago, being first recorded in Britain near Torquay in 1879! However it was not described from Britain until 1993, when it was known to have occurred since at least 1986 and 1989 as flourishing populations in Portsmouth (Hampshire) and Swanage (Dorset). There was also a population in Westcliff-on-Sea (Essex) recorded in 1990, and another in Littlehampton and Worthing (West Sussex). Its distribution is spreading more widely along the coast in the south and also inland, with confirmed records from South Devon, East Sussex, Kent, Surrey and Warwick. The large, grape-like individuals are the females and the smaller, more elongate ones, the males. These spiders are have become known as False Widows and, because of their colour, shape and size, are frequently mistaken for the Black Widow Spider that are found in warmer climes, but not in Britain (although some occasionally come into the country in packaged fruit and flowers). Black Widow Spiders belong to the world-wide genus Latrodectus. -

The Behavioural Ecology of Latrodectus Hasselti (Thorell), the Australian Redback Spider (Araneae: Theridiidae): a Review
Records of the Western Australian MIISellnl Supplement No. 52: 13-24 (1995). The behavioural ecology of Latrodectus hasselti (Thorell), the Australian Redback Spider (Araneae: Theridiidae): a review Lyn Forster McMasters Road, RD 1, Saddle Hill, Dunedin, New Zealand Abstract - Aspects of the biogeographical history and behavioural ecology of the AustralIan Latrodectus hasseIti provide support for the endemic status of this species. Cannibalism, prey stealing and short instar lengths are growth strategies for. female spiders whereas early maturation, small size, hiding and scavengmg are useful survival tactics for males. Moreover male complicity is an important component of sexual cannibalism which is ~hown to be a highly predictable event. Latrodectus hasseIti males hybridize with female L. katlpo (a New Zealand species) and fertile Fl and F2 generations Imply genetic relatedness. Hence, it is likely that L. hasselti and L. katipo evolv~d from a common ancestor in ancient Pangaea, a feasible explanation only If L. hasseItl IS endemic to Australia. It is concluded that L. hasseIti would have been able to persist in outback Australia for millions of years, with ItS mtraspeClfJc predatory habits aiding subsistence and the evolution of sexual cannibalism providing a way of coping with infrequent meeting and matmg opportunities. INTRODUCTION indigenous status, Main (1993) notes that, (as a Many stories and articles have been written consequence of its supposed introduction), "the about the redback spider (McKeown 1963; Raven absence of Latrodectus in the Australian region, 1992) with considerable attention being devoted to prior to human habitation, poses a curious its venomous nature (Southcott 1978; Sutherland zoogeographic dilemma". This comment raises an and Trinca 1978). -

Common Spiders in the Darwin Area D
Agnote No: I63 July 2014 Common Spiders in the Darwin Area D. Chin*, G. R. Brown*, T. Churchill2, J. Webber3 and H. Brown, Plant Industries, Darwin * Formerly DPIF 2 Formerly with the Tropical Ecosystems Research Centre, CSIRO, Darwin 3 Formerly with the CRC for Tropical Savannas Management, CDU, Darwin items that have been left undisturbed for long INTRODUCTION periods. The webs are loosely structured, strong and Spiders are invertebrate animals belonging to a have sticky basal strands. group called arachnids (which includes mites, ticks and scorpions). All spiders are predators and feed The female spiders rarely bite unless they are on insects, or other invertebrates, and may touched or handled. Although no fatalities due to sometimes capture small frogs or lizards. Spiders bites have been recorded since the introduction of have a variety of habits depending on where they anti-venom in 1956, bites are painful and must be live and how they feed. Some spiders build webs to treated as potentially dangerous. The male spider is capture flying insects while others may actively hunt much smaller and is not considered dangerous. for prey. Amongst ground-dwelling spiders, some Redbacks have a spherical abdomen, black legs live in burrows where they ambush crawling insects, and a black cephalothorax (where the legs are whereas others may hide under rocks and leaf litter attached). The female usually has a red stripe on the and search for prey at night. Spiders living on plants top side (dorsal side) of the abdomen and an have a variety of ways to catch insects and other hourglass shaped red mark on the underside prey and are useful in agriculture where they help in (ventral side) of the abdomen. -

Antivenoms for the Treatment of Spider Envenomation
† Antivenoms for the Treatment of Spider Envenomation Graham M. Nicholson1,* and Andis Graudins1,2 1Neurotoxin Research Group, Department of Heath Sciences, University of Technology, Sydney, New South Wales, Australia 2Departments of Emergency Medicine and Clinical Toxicology, Westmead Hospital, Westmead, New South Wales, Australia *Correspondence: Graham M. Nicholson, Ph.D., Director, Neurotoxin Research Group, Department of Heath Sciences, University of Technology, Sydney, P.O. Box 123, Broadway, NSW, 2007, Australia; Fax: 61-2-9514-2228; E-mail: Graham. [email protected]. † This review is dedicated to the memory of Dr. Struan Sutherland who’s pioneering work on the development of a funnel-web spider antivenom and pressure immobilisation first aid technique for the treatment of funnel-web spider and Australian snake bites will remain a long standing and life-saving legacy for the Australian community. ABSTRACT There are several groups of medically important araneomorph and mygalomorph spiders responsible for serious systemic envenomation. These include spiders from the genus Latrodectus (family Theridiidae), Phoneutria (family Ctenidae) and the subfamily Atracinae (genera Atrax and Hadronyche). The venom of these spiders contains potent neurotoxins that cause excessive neurotransmitter release via vesicle exocytosis or modulation of voltage-gated sodium channels. In addition, spiders of the genus Loxosceles (family Loxoscelidae) are responsible for significant local reactions resulting in necrotic cutaneous lesions. This results from sphingomyelinase D activity and possibly other compounds. A number of antivenoms are currently available to treat envenomation resulting from the bite of these spiders. Particularly efficacious antivenoms are available for Latrodectus and Atrax/Hadronyche species, with extensive cross-reactivity within each genera. -

Conservation Status of New Zealand Araneae (Spiders), 2020
2021 NEW ZEALAND THREAT CLASSIFICATION SERIES 34 Conservation status of New Zealand Araneae (spiders), 2020 Phil J. Sirvid, Cor J. Vink, Brian M. Fitzgerald, Mike D. Wakelin, Jeremy Rolfe and Pascale Michel Cover: A large sheetweb sider, Cambridgea foliata – Not Threatened. Photo: Jeremy Rolfe. New Zealand Threat Classification Series is a scientific monograph series presenting publications related to the New Zealand Threat Classification System (NZTCS). Most will be lists providing NZTCS status of members of a plant or animal group (e.g. algae, birds, spiders). There are currently 23 groups, each assessed once every 5 years. From time to time the manual that defines the categories, criteria and process for the NZTCS will be reviewed. Publications in this series are considered part of the formal international scientific literature. This report is available from the departmental website in pdf form. Titles are listed in our catalogue on the website, refer www.doc.govt.nz under Publications. The NZTCS database can be accessed at nztcs.org.nz. For all enquiries, email [email protected]. © Copyright August 2021, New Zealand Department of Conservation ISSN 2324–1713 (web PDF) ISBN 978–1–99–115291–6 (web PDF) This report was prepared for publication by Te Rōpū Ratonga Auaha, Te Papa Atawhai/Creative Services, Department of Conservation; editing and layout by Lynette Clelland. Publication was approved by the Director, Terrestrial Ecosystems Unit, Department of Conservation, Wellington, New Zealand Published by Department of Conservation Te Papa Atawhai, PO Box 10420, Wellington 6143, New Zealand. This work is licensed under the Creative Commons Attribution 4.0 International licence. -

Safe Laboratory Management for Arachnids of Medical Importance
August 2011 Animal Technology and Welfare Safe laboratory management for arachnids of medical importance SARAH REED, MIKAELLA LOCK and STEVEN TRIM* Venomtech Ltd, PO Box 468, Ramsgate, Kent CT11 1BD *Corresponding author [email protected] (Presented at Institute of Animal Technology Congress 2011) Summary understanding of the species’ “normal” behaviour and Safe animal management is key to any facility; this is frequent, careful observation to identify any deviation especially true of those housing animals of significant from that normal behaviour. In addition, the husbandry medical importance. Procedures detailed here of arachnids of medical importance poses more demonstrate how dangerous arachnids including significant challenges in containment and safe handling scorpions (Family Buthidae ), wandering spiders (Family (for both animal and technician) due to the size, speed, Ctenidae ), recluse spiders (Family Sicariidae ) and the aggression and venomous characteristics of many Theridiidae family containing the widow spiders can be species. safely managed in the laboratory environment. This dramatically reduces the risk of envenomation during This paper describes a method of performing routine husbandry and experimental procedures. Risks are husbandry by which risk of envenomation is reduced with modified enclosures that allow separation significantly reduced along with risk of escape or injury from the animals during most husbandry procedures of the animals and was originally presented as a poster and use of anaesthesia where contact is inevitable. at the Laboratory Animal Science Association (LASA) The equipment and techniques presented here thus Winter Meeting and an oral presentation at the Institute allow for greatly improved safety when working with of Animal Technology (IAT) Congress 2011. -

The Second Redback Antivenom Evaluation (RAVE-II) Study
TOXICOLOGY/ORIGINAL RESEARCH Randomized Controlled Trial of Intravenous Antivenom Versus Placebo for Latrodectism: The Second Redback Antivenom Evaluation (RAVE-II) Study Geoffrey K. Isbister, FACEM, MD*; Colin B. Page, MBChB, FACEM; Nicholas A. Buckley, FRACP, MD; Daniel M. Fatovich, MBBS, FACEM; Ovidiu Pascu, MBBS, FACEM; Stephen P. J. MacDonald, MBChB, FACEM; Leonie A. Calver; Simon G. A. Brown, FACEM, PhD; on behalf of the RAVE Investigators *Corresponding Author. E-mail: [email protected]. Study objective: Latrodectism is the most important spider envenomation syndrome worldwide. There remains considerable controversy over antivenom treatment. We aimed to investigate whether antivenom resulted in resolution of pain and systemic effects in patients with latrodectism who received standardized analgesia. Methods: In a multicenter randomized placebo-controlled trial of redback spider antivenom for latrodectism, 224 patients (>7 years) with a redback spider bite and severe pain, with or without systemic effects, were randomized to receive normal saline solution (placebo) or antivenom after receiving standardized analgesia. The primary outcome was a clinically significant reduction in pain 2 hours after trial medication compared with baseline. A second primary outcome for the subgroup with systemic features of envenomation was resolution of systemic features at 2 hours. Secondary outcomes were improved pain at 4 and 24 hours, resolution of systemic features at 4 hours, administration of opioid analgesics or unblinded antivenom after 2 hours, and adverse reactions. Results: Two hours after treatment, 26 of 112 patients (23%) from the placebo arm had a clinically significant improvement in pain versus 38 of 112 (34%) from the antivenom arm (difference in favor of antivenom 10.7%; 95% confidence interval 1.1% to 22.6%; P .10). -

Journal of Toxicologic Pathology
Advance Publication Journal of Toxicologic Pathology Received : September 14, 2017 Accepted : December 19, 2017 J-STAGE Advance Published Date : February 19, 2018 Japanese Society of Toxicologic Pathology Freeze-dried equine-derived redback spider antivenom: A local irritation study by intramuscular injection in rabbits and a repeated-dose toxicity study in rats Akihiko Yamamoto1, Satomi Harano2, Noriko Shinya2, Ayataka Nagano2, Yoshinobu Miyatsu2, Kyouko Sawabe3, Takayuki Matsumura4, Manabu Ato4, Motohide Takahashi5, Hisashi Taki6 and Toru Hifumi7 1Division of Biosafety Control and Research, National Institute of Infectious Disease, 4-7-1 Gakuen, Musashimurayama-shi, Tokyo 208-0011, Japan 2The Chemo-Sero-Therapeutic Research Institute (KAKETSUKEN), 1-6-1 Okubo, Kita-ku, Kumamoto-shi, Kumamoto 860-8568, Japan 3Department of Medical Entomology, National Institute of Infectious Diseases, 1-23-1 Toyama, Shinjuku-ku, Tokyo 162-8640, Japan 4Department of Immunology, National Institute of Infectious Disease, 1-23-1 Toyama, Shinjuku-ku, Tokyo 162-8640, Japan 5Pharmaceuticals and Medical Devices Agency, 3-3-2 Kasumigaseki, Chiyoda-ku, Tokyo 100-0013 Japan 1 6Ministry of Health, Labour and Welfare, Tokyo 100-8916, Japan 7Emergency Medical Center, Kagawa University Hospital, 1750-1 Ikenobe, Miki, Kita, Kagawa 761-0793, Japan Corresponding author: Akihiko Yamamoto, Division of Biosafety Control and Research, National Institute of Infectious Disease, 4-7-1 Gakuen, Musashimurayama-shi, Tokyo 208-0011, Japan Tel: +81-42-561-0771 Ext. 3734 Fax: +81-42-564-4881 E-mail: [email protected] 2 Abstract The redback spider (Latrodectus hasseltii) is nonindigenous to Japan but has now spread throughout the country. Bites to humans are rare but can be fatal. -

Health Tips Factsheets
HEALTH TIPS FACTSHEETS How to stay healthy at Cuboree HEALTH TIPS! #1 Washing your hands – visibly dirty Wash your hands with soap and water when they are visibly soiled. Use a hand sanitiser afterwards…! MORE INFORMATION! For more information, visit your Sub Camp HQ HEALTH TIPS! #1 Washing your hands – visibly dirty – detailed information. Why it’s important A number of infectious diseases can be spread from one person to another by contaminated hands, particularly gastrointestinal infections and hepatitis A. Washing your hands properly can help prevent the spread of the organisms that cause these diseases. Some forms of gastroenteritis can cause serious complications, especially for young children or those with a weakened immune system. Drying your hands properly is as important as washing them. When to wash your hands You should wash your hands thoroughly: • Before preparing food • Before eating • Between handling raw and cooked or ready-to-eat food • After going to the toilet • After using a tissue or handkerchief • After handling rubbish How to wash your hands properly To wash hands properly: • Wet your hands with warm water. • Apply one dose of liquid soap and lather well for 15–20 seconds (or longer if the dirt is ingrained). • Rub hands together rapidly across all surfaces of your hands and wrists to help remove dirt and germs. • Don’t forget the backs of your hands, your wrists, between your fingers and under your fingernails. • Wash your hands for at least 10 to 15 seconds. • Rinse well under running water and make sure all traces of soap are removed, as residues may cause irritation. -

Otago-Cms-2016-Volume-1.Pdf
CMS CONSERVATION MANAGEMENT STRATEGY Otago 2016, Volume 1 CMS CONSERVATION MANAGEMENT STRATEGY Otago 2016, Volume 1 Cover image: Celmisia haastii on the upper Wye valley near Queenstown with the Tāpuae-o-Uenuku/Hector Mountains in the background. Photo: John Barkla September 2016, New Zealand Department of Conservation ISBN 978-0-478-15088-9 (print) ISBN 978-0-478-15089-6 (online) Crown copyright © 2016 This work is licensed under the Creative Commons Attribution 4.0 International licence. In essence, you are free to copy, distribute and adapt the work, as long as you attribute the work to the Crown and abide by the other licence terms. To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/. Please note that no departmental or governmental emblem, logo or Coat of Arms may be used in any way which infringes any provision of the Flags, Emblems, and Names Protection Act 1981. Attribution to the Crown should be in written form and not by reproduction of any such emblem, logo or Coat of Arms. Use the wording ‘Department of Conservation’ in your attribution, not the Department of Conservation logo. This publication is produced using paper sourced from well-managed, renewable and legally logged forests. Contents Foreword 7 Introduction 8 Purpose of conservation management strategies 8 Treaty partnership with Ngāi Tahu 9 CMS structure 11 Interpretation 11 CMS term 12 Relationship with other Department of Conservation strategic documents and tools 12 Relationships with other planning processes 14 The Department’s legislative -
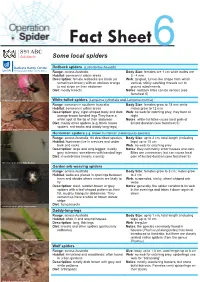
Some Local Australian Spiders • Fact Sheet
Fact Sheet6 Some local spiders Redback spiders (Latrodectus hasselti) Range: across Australia Body Size: females are 1 cm while males are Habitat: common in urban areas 3 - 4 mm Description: female redbacks are black (or Web: tangled, funnel-like shape from which sometimes brown) with an obvious orange vertical, sticky catching threads run to to red stripe on their abdomen ground attachments Diet: mostly insects Notes: redback bites can be serious (see factsheet 5) Redback spider; photo: John Hodgson White-tailed spiders (Lampona cylindrata and Lampona murina) Range: common in southern Australia Body Size: females grow to 18 mm while Habitat: common in urban areas males grow to 12 mm Description: grey, cigar-shaped body and dark Web: no web for catching prey, they hunt at orange-brown banded legs They have a night white spot at the tip of their abdomen Notes: white-tail bites cause local pain of Diet: mainly other spiders (e.g. black house limited duration (see factsheet 5) spiders, red-backs and daddy-long-legs) White-tailed spider; photo: John Hodgson Huntsman spiders e.g. brown huntsman (Heteropoda species) Range: across Australia; 94 described species, Body Size: up to 2 cm; total length (including Habitat: huntsmen live in crevices and under legs) up to 15 cm bark and rocks Web: no web for catching prey Description: large and long-legged; mostly Notes: they commonly enter houses and cars. grey to brown, sometimes with banded legs Bites are uncommon, but can cause local Diet: invertebrates (mainly insects) pain of limited duration (see factsheet 5) Huntsman spider; photo: Philip Roetman Garden orb weaving spiders Range: across Australia Body Size: females grow to 3 cm; males grow Habitat: webs are placed in openings between to 2 cm trees and shrubs where insects are likely to Web: suspended, sticky, wheel-shaped orb fly webs Description: stout, reddish-brown or grey Notes: generally, the spider constructs its web spiders with a leaf-shaped pattern on their in the evenings and takes it down again at fat, roughly triangular abdomens. -

The Efficacy of Antivenin Latrodectus (Black Widow) Equine Immune F
TOXICOLOGY/ORIGINAL RESEARCH The Efficacy of Antivenin Latrodectus (Black ʹ Widow) Equine Immune F(ab )2 Versus Placebo in the Treatment of Latrodectism: A Randomized, Double-Blind, Placebo-Controlled, Clinical Trial Richard C. Dart, MD, PhD*; Sean P. Bush, MD; Kennon Heard, MD, PhD; Thomas C. Arnold, MD; Mark Sutter, MD; Danielle Campagne, MD; Christopher P. Holstege, MD; Steven A. Seifert, MD; Jean C. Y. Lo, MD; Dan Quan, DO; Stephen Borron, MD; David A. Meurer, MD; Randy I. Burnham, MS; Jude McNally, PharmD; Walter Garcia-Ubbelohde, MD; Victoria E. Anderson, MPH *Corresponding Author. E-mail: [email protected]. Study objective: The antivenom currently available for treatment of systemic black widow envenomation (latrodectism) is composed of equine whole immunoglobin. Although considered effective, it has been associated with anaphylaxis and 2 reported fatalities. We test the efficacy and safety of new equine antivenom composed of purified F(abʹ)2 antibody fragments. Methods: A randomized, double-blind, placebo-controlled trial was conducted at 16 sites across the United States. Subjects aged 10 years or older with moderate to severe pain because of black widow spider envenomation received F(abʹ)2 antivenom or placebo. The primary outcome measure was treatment failure, which was defined as failure to achieve and maintain clinically significant reduction in pain for 48 hours posttreatment. Secondary measures of pain intensity differences and summed pain intensity difference were computed. Adverse events were recorded. Results: Sixty patients were treated (29 antivenom and 31 placebo). The mean age was 39 years and 68% were male. There were 15 treatment failures in the antivenom group and 24 in the placebo group (P¼.019).Differencesinpainintensity difference between groups were lower at each postbaseline point, and the mean summed pain intensity difference was greater for the antivenom group (difference 2,133; 95% confidence interval 177 to 4,090).