Computational Quantum Chemistry Tools.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021](https://docslib.b-cdn.net/cover/7378/automated-construction-of-quantum-classical-hybrid-models-arxiv-2102-09355v1-physics-chem-ph-18-feb-2021-177378.webp)
Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021
Automated construction of quantum{classical hybrid models Christoph Brunken and Markus Reiher∗ ETH Z¨urich, Laboratorium f¨urPhysikalische Chemie, Vladimir-Prelog-Weg 2, 8093 Z¨urich, Switzerland February 18, 2021 Abstract We present a protocol for the fully automated construction of quantum mechanical-(QM){ classical hybrid models by extending our previously reported approach on self-parametri- zing system-focused atomistic models (SFAM) [J. Chem. Theory Comput. 2020, 16 (3), 1646{1665]. In this QM/SFAM approach, the size and composition of the QM region is evaluated in an automated manner based on first principles so that the hybrid model describes the atomic forces in the center of the QM region accurately. This entails the au- tomated construction and evaluation of differently sized QM regions with a bearable com- putational overhead that needs to be paid for automated validation procedures. Applying SFAM for the classical part of the model eliminates any dependence on pre-existing pa- rameters due to its system-focused quantum mechanically derived parametrization. Hence, QM/SFAM is capable of delivering a high fidelity and complete automation. Furthermore, since SFAM parameters are generated for the whole system, our ansatz allows for a con- venient re-definition of the QM region during a molecular exploration. For this purpose, a local re-parametrization scheme is introduced, which efficiently generates additional clas- sical parameters on the fly when new covalent bonds are formed (or broken) and moved to the classical region. arXiv:2102.09355v1 [physics.chem-ph] 18 Feb 2021 ∗Corresponding author; e-mail: [email protected] 1 1 Introduction In contrast to most protocols of computational quantum chemistry that consider isolated molecules, chemical processes can take place in a vast variety of complex environments. -

Spoken Tutorial Project, IIT Bombay Brochure for Chemistry Department
Spoken Tutorial Project, IIT Bombay Brochure for Chemistry Department Name of FOSS Applications Employability GChemPaint GChemPaint is an editor for 2Dchem- GChemPaint is currently being developed ical structures with a multiple docu- as part of The Chemistry Development ment interface. Kit, and a Standard Widget Tool kit- based GChemPaint application is being developed, as part of Bioclipse. Jmol Jmol applet is used to explore the Jmol is a free, open source molecule viewer structure of molecules. Jmol applet is for students, educators, and researchers used to depict X-ray structures in chemistry and biochemistry. It is cross- platform, running on Windows, Mac OS X, and Linux/Unix systems. For PG Students LaTeX Document markup language and Value addition to academic Skills set. preparation system for Tex typesetting Essential for International paper presentation and scientific journals. For PG student for their project work Scilab Scientific Computation package for Value addition in technical problem numerical computations solving via use of computational methods for engineering problems, Applicable in Chemical, ECE, Electrical, Electronics, Civil, Mechanical, Mathematics etc. For PG student who are taking Physical Chemistry Avogadro Avogadro is a free and open source, Research and Development in Chemistry, advanced molecule editor and Pharmacist and University lecturers. visualizer designed for cross-platform use in computational chemistry, molecular modeling, material science, bioinformatics, etc. Spoken Tutorial Project, IIT Bombay Brochure for Commerce and Commerce IT Name of FOSS Applications / Employability LibreOffice – Writer, Calc, Writing letters, documents, creating spreadsheets, tables, Impress making presentations, desktop publishing LibreOffice – Base, Draw, Managing databases, Drawing, doing simple Mathematical Math operations For Commerce IT Students Drupal Drupal is a free and open source content management system (CMS). -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -
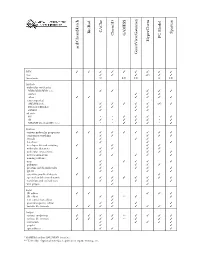
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Pymol Modelling Workshop
PyMOL Modelling Workshop My website: http://pldserver1.biochem.queensu.ca/~rlc/work/teaching/BCHM442/ There you will find links to download the latest educational version of PyMOL as well as a link to my “Introduction to PyMOL”, which in turn has links to other people's PyMOL tutorials. Note also the PyMOL Wiki: http://pymolwiki.org. Structure files can be found by searching the Protein Data Bank (PDB) for structure: easy to remember website http://www.pdb.org. There is also the PDBe (PDB Europe, http://www.pdbe.org) that contains the same databank of structures, but with a different web interface for searching for structures and a different set of tools for analyzing structures. What is in a PDB file? Lots of information in the “header” (the section of the file preceding the actual atomic coordinates) as well as the coordinates for the atoms. When assessing a structure, one needs to take account of the resolution and R-factor, error estimates and missing residues. There is information about the sequence that was used to determine the structure with a sequence database reference. There is also information about the biological unit. In the case of crystal structures the biological unit may need to be generated by applying crystallographic symmetry operators, although there are web sites that also try to provide that information. (e.g. http://www.ebi.ac.uk/msd-srv/pisa/). Warning: One cannot blindly trust a crystal structure to be providing you with a completely accurate picture of reality. Reading the paper that describes the structure is a good start! Outline of PyMOL usage PyMOL is a very powerful, scriptable (customizable) tool for making publication-quality figures and performing analyses on structures. -

Dmol Guide to Select a Dmol3 Task 1
DMOL3 GUIDE MATERIALS STUDIO 8.0 Copyright Notice ©2014 Dassault Systèmes. All rights reserved. 3DEXPERIENCE, the Compass icon and the 3DS logo, CATIA, SOLIDWORKS, ENOVIA, DELMIA, SIMULIA, GEOVIA, EXALEAD, 3D VIA, BIOVIA and NETVIBES are commercial trademarks or registered trademarks of Dassault Systèmes or its subsidiaries in the U.S. and/or other countries. All other trademarks are owned by their respective owners. Use of any Dassault Systèmes or its subsidiaries trademarks is subject to their express written approval. Acknowledgments and References To print photographs or files of computational results (figures and/or data) obtained using BIOVIA software, acknowledge the source in an appropriate format. For example: "Computational results obtained using software programs from Dassault Systèmes Biovia Corp.. The ab initio calculations were performed with the DMol3 program, and graphical displays generated with Materials Studio." BIOVIA may grant permission to republish or reprint its copyrighted materials. Requests should be submitted to BIOVIA Support, either through electronic mail to [email protected], or in writing to: BIOVIA Support 5005 Wateridge Vista Drive, San Diego, CA 92121 USA Contents DMol3 1 Setting up a molecular dynamics calculation20 Introduction 1 Choosing an ensemble 21 Further Information 1 Defining the time step 21 Tasks in DMol3 2 Defining the thermostat control 21 Energy 3 Constraints during dynamics 21 Setting up the calculation 3 Setting up a transition state calculation 22 Dynamics 4 Which method to use? -

Atomistic Molecular Dynamics Simulation of Graphene- Isoprene Nanocomposites
Atomistic molecular dynamics simulation of graphene- isoprene nanocomposites P Chanlert1, J Wong-Ekkabut2, W Liewrian3,4,5 and T Sutthibutpong3,4,5* 1 Program of Physics, Faculty of Science and Technology, Songkhla Rajabhat University, Songkhla, 90000, Thailand 2 Department of Physics, Faculty of Science, Kasetsart University, Bangkok 10900, Thailand 3 Theoretical and Computational Physics Group, Department of Physics, Faculty of Science, King Mongkut’s University of Technology Thonburi (KMUTT), Bangkok 10140, Thailand 4 Theoretical and Computational Science Center (TaCS), Science Laboratory Building, Faculty of Science, King Mongkut’s University of Technology Thonburi (KMUTT), Bangkok 10140, Thailand 5 Thailand Center of Excellence in Physics (ThEP Center), Commission on Higher Education, Bangkok 10400, Thailand *email: [email protected] Abstract. A series of atomistic molecular dynamics simulations were performed for 4-mer isoprene molecules confined between graphene sheets with varied graphene separations in order to observe the finite size effects arose from the van der Waals interactions between the isoprene oligomer, forming high density shells at the graphene interfaces. For the small confinement regions, 1.24 nm and 2.21 nm, local density of isoprene at the graphene interface becomes higher than 10 times the bulk density. These results provided the further insights towards the rational design of nanostructures based on graphene sheets and natural rubber polymer. 1. Introduction Composite material is the combination between two materials or more where chemical reaction between these materials is absent. In composites, one with higher quantity is called matrix while those with lesser quantity are called fillers. In general, adding fillers into matrix material enhances both physical and mechanical properties in some ways. -
![Modern Quantum Chemistry with [Open]Molcas](https://docslib.b-cdn.net/cover/4742/modern-quantum-chemistry-with-open-molcas-744742.webp)
Modern Quantum Chemistry with [Open]Molcas
Modern quantum chemistry with [Open]Molcas Cite as: J. Chem. Phys. 152, 214117 (2020); https://doi.org/10.1063/5.0004835 Submitted: 17 February 2020 . Accepted: 11 May 2020 . Published Online: 05 June 2020 Francesco Aquilante , Jochen Autschbach , Alberto Baiardi , Stefano Battaglia , Veniamin A. Borin , Liviu F. Chibotaru , Irene Conti , Luca De Vico , Mickaël Delcey , Ignacio Fdez. Galván , Nicolas Ferré , Leon Freitag , Marco Garavelli , Xuejun Gong , Stefan Knecht , Ernst D. Larsson , Roland Lindh , Marcus Lundberg , Per Åke Malmqvist , Artur Nenov , Jesper Norell , Michael Odelius , Massimo Olivucci , Thomas B. Pedersen , Laura Pedraza-González , Quan M. Phung , Kristine Pierloot , Markus Reiher , Igor Schapiro , Javier Segarra-Martí , Francesco Segatta , Luis Seijo , Saumik Sen , Dumitru-Claudiu Sergentu , Christopher J. Stein , Liviu Ungur , Morgane Vacher , Alessio Valentini , and Valera Veryazov J. Chem. Phys. 152, 214117 (2020); https://doi.org/10.1063/5.0004835 152, 214117 © 2020 Author(s). The Journal ARTICLE of Chemical Physics scitation.org/journal/jcp Modern quantum chemistry with [Open]Molcas Cite as: J. Chem. Phys. 152, 214117 (2020); doi: 10.1063/5.0004835 Submitted: 17 February 2020 • Accepted: 11 May 2020 • Published Online: 5 June 2020 Francesco Aquilante,1,a) Jochen Autschbach,2,b) Alberto Baiardi,3,c) Stefano Battaglia,4,d) Veniamin A. Borin,5,e) Liviu F. Chibotaru,6,f) Irene Conti,7,g) Luca De Vico,8,h) Mickaël Delcey,9,i) Ignacio Fdez. Galván,4,j) Nicolas Ferré,10,k) Leon Freitag,3,l) Marco Garavelli,7,m) Xuejun Gong,11,n) Stefan Knecht,3,o) Ernst D. Larsson,12,p) Roland Lindh,4,q) Marcus Lundberg,9,r) Per Åke Malmqvist,12,s) Artur Nenov,7,t) Jesper Norell,13,u) Michael Odelius,13,v) Massimo Olivucci,8,14,w) Thomas B. -

Benchmarking and Application of Density Functional Methods In
BENCHMARKING AND APPLICATION OF DENSITY FUNCTIONAL METHODS IN COMPUTATIONAL CHEMISTRY by BRIAN N. PAPAS (Under Direction the of Henry F. Schaefer III) ABSTRACT Density Functional methods were applied to systems of chemical interest. First, the effects of integration grid quadrature choice upon energy precision were documented. This was done through application of DFT theory as implemented in five standard computational chemistry programs to a subset of the G2/97 test set of molecules. Subsequently, the neutral hydrogen-loss radicals of naphthalene, anthracene, tetracene, and pentacene and their anions where characterized using five standard DFT treatments. The global and local electron affinities were computed for the twelve radicals. The results for the 1- naphthalenyl and 2-naphthalenyl radicals were compared to experiment, and it was found that B3LYP appears to be the most reliable functional for this type of system. For the larger systems the predicted site specific adiabatic electron affinities of the radicals are 1.51 eV (1-anthracenyl), 1.46 eV (2-anthracenyl), 1.68 eV (9-anthracenyl); 1.61 eV (1-tetracenyl), 1.56 eV (2-tetracenyl), 1.82 eV (12-tetracenyl); 1.93 eV (14-pentacenyl), 2.01 eV (13-pentacenyl), 1.68 eV (1-pentacenyl), and 1.63 eV (2-pentacenyl). The global minimum for each radical does not have the same hydrogen removed as the global minimum for the analogous anion. With this in mind, the global (or most preferred site) adiabatic electron affinities are 1.37 eV (naphthalenyl), 1.64 eV (anthracenyl), 1.81 eV (tetracenyl), and 1.97 eV (pentacenyl). In later work, ten (scandium through zinc) homonuclear transition metal trimers were studied using one DFT 2 functional. -

Quantum Mechanical Calculation of Molybdenum and Tungsten Influence on the Crm-Oxide Catalyst Acidity Oyegoke Toyese1 Fadimatu N
Hittite Journal of Science and Engineering, 2020, 7 (4) 297–311 ISSN NUMBER: 2148–4171 DOI: 10.17350/HJSE19030000199 Quantum Mechanical Calculation of Molybdenum and Tungsten Influence on the CrM-oxide Catalyst Acidity Oyegoke Toyese1 Fadimatu N. Dabai1 Adamu Uzairur2 and Baba El-Yakubu Jibril1 1Ahmadu Bello University, Department of Chemical Engineering, Zaria, Nigeria 2Ahmadu Bello University, Department of Chemistry, Zaria, Nigeria Article History: ABSTRACT Received: 2020/06/18 Accepted: 2020/11/02 Online: 2020/12/31 emi-empirical calculations were employed to understand the effects of introducing pro- Smoters such as molybdenum (Mo) and tungsten (W) on chromium (III) oxide catalyst Correspondence to: Oyegoke Toyese, for the dehydrogenation of propane into propylene. For this purpose, we investigated CrM- Ahmadu Bello University, Chemical Engineering, Zaria, NIGERIA oxide (M = Cr, Mo, and W) catalysts. In this study, the Lewis acidity of the catalyst was ex- E-Mail: [email protected] amined using Lewis acidity parameters (Ac), including ammonia and pyridine adsorption Phone: +234 703 047 91 06 energy. The results obtained from this study of overall acidity across all sites of the catalysts studied reveal Mo-modified catalyst as the one with the least acidity while the W-modified catalyst was found to have shown the highest acidity signifies that the introduction of Mo would reduce acidity while W accelerates it. The finding, therefore, confirms tungsten (W) to be more influential and would be more promising when compared to molybdenum (Mo) due to the better avenue that is offered by W for the promotion of electron exchange and its higher acidity(s). -

1Jwsn As a Solution State Predictor for Tungsten-Tin Solid State Bond
Organometallics 2005, 24, 3897-3906 3897 1 JWSn as a Solution State Predictor for Tungsten-Tin Solid State Bond Length in Sterically Crowded Tungstenocene Stannyl Complexes T. Andrew Mobley,* Rochelle Gandour, Eric P. Gillis, Kwame Nti-Addae, Rahul Palchaudhuri, Presha Rajbhandari, Neil Tomson, Angel Vargas, and Qi Zheng Noyce Science Center, Grinnell College, 1116 8th Street, Grinnell, Iowa 50112 Received December 14, 2004 A correlation between the tungsten tin solid state bond distance (rWSn) and the solution 1 state one-bond scalar coupling of tungsten to tin ( JWSn) is reported. This correlation was observed in hydrido- and chlorotungstenocene diorganostannyl chlorides (Cp2WXSnClR2,X ) H(1), X ) Cl (2)) with sterically demanding organic substituents (R ) tBu (1a, 2a), Ph (1b, 2b)). The different steric demands of the organic substituents (tBu and Ph) force the complexes 1a and 1b (or 2a and 2b) to adopt at 190 K in the solid state different, single rotameric conformations. For 1a and 2a, the tBu substituents occupy the equatorial wedge of the tungstenocene, whereas for 1b and 2b the Ph substituents are rotated to place the chlorine in the equatorial wedge. Variable-temperature multinuclear NMR and density functional theory calculations at the B3LYP/LANL2DZ level indicate the possibility of low- lying conformational isomers for complexes 1b. Introduction solid and solution state structures of stannyl complexes with both small (Me) and more sterically hindered (t- Mid-transition metal stannyl complexes represent an Bu, CH(SiMe3)2) organic substituents. In the process of 1-9 area of increasing interest due to their potential as exploring the reactive chemistry of transition metal 8,9 unique reagents and catalysts. -

A Summary of ERCAP Survey of the Users of Top Chemistry Codes
A survey of codes and algorithms used in NERSC chemical science allocations Lin-Wang Wang NERSC System Architecture Team Lawrence Berkeley National Laboratory We have analyzed the codes and their usages in the NERSC allocations in chemical science category. This is done mainly based on the ERCAP NERSC allocation data. While the MPP hours are based on actually ERCAP award for each account, the time spent on each code within an account is estimated based on the user’s estimated partition (if the user provided such estimation), or based on an equal partition among the codes within an account (if the user did not provide the partition estimation). Everything is based on 2007 allocation, before the computer time of Franklin machine is allocated. Besides the ERCAP data analysis, we have also conducted a direct user survey via email for a few most heavily used codes. We have received responses from 10 users. The user survey not only provide us with the code usage for MPP hours, more importantly, it provides us with information on how the users use their codes, e.g., on which machine, on how many processors, and how long are their simulations? We have the following observations based on our analysis. (1) There are 48 accounts under chemistry category. This is only second to the material science category. The total MPP allocation for these 48 accounts is 7.7 million hours. This is about 12% of the 66.7 MPP hours annually available for the whole NERSC facility (not accounting Franklin). The allocation is very tight. The majority of the accounts are only awarded less than half of what they requested for.