Scale QM/MM-MD Simulations and Its Application to Biomolecular Systems
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021](https://docslib.b-cdn.net/cover/7378/automated-construction-of-quantum-classical-hybrid-models-arxiv-2102-09355v1-physics-chem-ph-18-feb-2021-177378.webp)
Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021
Automated construction of quantum{classical hybrid models Christoph Brunken and Markus Reiher∗ ETH Z¨urich, Laboratorium f¨urPhysikalische Chemie, Vladimir-Prelog-Weg 2, 8093 Z¨urich, Switzerland February 18, 2021 Abstract We present a protocol for the fully automated construction of quantum mechanical-(QM){ classical hybrid models by extending our previously reported approach on self-parametri- zing system-focused atomistic models (SFAM) [J. Chem. Theory Comput. 2020, 16 (3), 1646{1665]. In this QM/SFAM approach, the size and composition of the QM region is evaluated in an automated manner based on first principles so that the hybrid model describes the atomic forces in the center of the QM region accurately. This entails the au- tomated construction and evaluation of differently sized QM regions with a bearable com- putational overhead that needs to be paid for automated validation procedures. Applying SFAM for the classical part of the model eliminates any dependence on pre-existing pa- rameters due to its system-focused quantum mechanically derived parametrization. Hence, QM/SFAM is capable of delivering a high fidelity and complete automation. Furthermore, since SFAM parameters are generated for the whole system, our ansatz allows for a con- venient re-definition of the QM region during a molecular exploration. For this purpose, a local re-parametrization scheme is introduced, which efficiently generates additional clas- sical parameters on the fly when new covalent bonds are formed (or broken) and moved to the classical region. arXiv:2102.09355v1 [physics.chem-ph] 18 Feb 2021 ∗Corresponding author; e-mail: [email protected] 1 1 Introduction In contrast to most protocols of computational quantum chemistry that consider isolated molecules, chemical processes can take place in a vast variety of complex environments. -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -
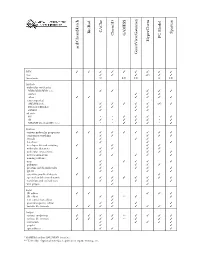
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Dr. David Danovich
Dr. David Danovich ! 14 January 2021 (h-index: 34; 124 publications) Personal Information Name: Contact information Danovich David Private: Nissan Harpaz st., 4/9, Jerusalem, Date of Birth: 9371643 Israel June 29, 1959 Mobile: +972-54-4768669 E-mail: [email protected] Place of Birth: Irkutsk, USSR Work: Institute of Chemistry, The Hebrew Citizenship: University, Edmond Safra Campus, Israeli Givat Ram, 9190401 Jerusalem, Israel Marital status: Phone: +972-2-6586934 Married +2 FAX: +972-2-6584033 E-mail: [email protected] Current position 1992- present: Senior computational chemist and scientific programmer in the Institute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, Israel Scientific skills Very experienced scientific researcher with demonstrated ability to implement ab initio molecular electronic structure and valence bond calculations for obtaining insights into physical and chemical properties of molecules and materials. Extensive expertise in implementation of Molecular Orbital Theory based on ab initio methods such as Hartree-Fock theory (HF), Density Functional Theory (DFT), Moller-Plesset Perturbation theory (MP2-MP5), Couple Cluster theories [CCST(T)], Complete Active Space (CAS, CASPT2) theories, Multireference Configuration Interaction (MRCI) theory and modern ab initio valence bond theory (VBT) methods such as Valence Bond Self Consistent Field theory (VBSCF), Breathing Orbital Valence Bond theory (BOVB) and its variants (like D-BOVB, S-BOVB and SD-BOVB), Valence Bond Configuration Interaction theory (VBCI), Valence Bond Perturbation theory (VBPT). Language skills Advanced Conversational English, Russian Hebrew Academic background 1. Undergraduate studies, 1978-1982 Master of Science in Physics, June 1982 Irkutsk State University, USSR Dr. David Danovich ! 2. Graduate studies, 1984-1989 Ph. -
![Modern Quantum Chemistry with [Open]Molcas](https://docslib.b-cdn.net/cover/4742/modern-quantum-chemistry-with-open-molcas-744742.webp)
Modern Quantum Chemistry with [Open]Molcas
Modern quantum chemistry with [Open]Molcas Cite as: J. Chem. Phys. 152, 214117 (2020); https://doi.org/10.1063/5.0004835 Submitted: 17 February 2020 . Accepted: 11 May 2020 . Published Online: 05 June 2020 Francesco Aquilante , Jochen Autschbach , Alberto Baiardi , Stefano Battaglia , Veniamin A. Borin , Liviu F. Chibotaru , Irene Conti , Luca De Vico , Mickaël Delcey , Ignacio Fdez. Galván , Nicolas Ferré , Leon Freitag , Marco Garavelli , Xuejun Gong , Stefan Knecht , Ernst D. Larsson , Roland Lindh , Marcus Lundberg , Per Åke Malmqvist , Artur Nenov , Jesper Norell , Michael Odelius , Massimo Olivucci , Thomas B. Pedersen , Laura Pedraza-González , Quan M. Phung , Kristine Pierloot , Markus Reiher , Igor Schapiro , Javier Segarra-Martí , Francesco Segatta , Luis Seijo , Saumik Sen , Dumitru-Claudiu Sergentu , Christopher J. Stein , Liviu Ungur , Morgane Vacher , Alessio Valentini , and Valera Veryazov J. Chem. Phys. 152, 214117 (2020); https://doi.org/10.1063/5.0004835 152, 214117 © 2020 Author(s). The Journal ARTICLE of Chemical Physics scitation.org/journal/jcp Modern quantum chemistry with [Open]Molcas Cite as: J. Chem. Phys. 152, 214117 (2020); doi: 10.1063/5.0004835 Submitted: 17 February 2020 • Accepted: 11 May 2020 • Published Online: 5 June 2020 Francesco Aquilante,1,a) Jochen Autschbach,2,b) Alberto Baiardi,3,c) Stefano Battaglia,4,d) Veniamin A. Borin,5,e) Liviu F. Chibotaru,6,f) Irene Conti,7,g) Luca De Vico,8,h) Mickaël Delcey,9,i) Ignacio Fdez. Galván,4,j) Nicolas Ferré,10,k) Leon Freitag,3,l) Marco Garavelli,7,m) Xuejun Gong,11,n) Stefan Knecht,3,o) Ernst D. Larsson,12,p) Roland Lindh,4,q) Marcus Lundberg,9,r) Per Åke Malmqvist,12,s) Artur Nenov,7,t) Jesper Norell,13,u) Michael Odelius,13,v) Massimo Olivucci,8,14,w) Thomas B. -

Benchmarking and Application of Density Functional Methods In
BENCHMARKING AND APPLICATION OF DENSITY FUNCTIONAL METHODS IN COMPUTATIONAL CHEMISTRY by BRIAN N. PAPAS (Under Direction the of Henry F. Schaefer III) ABSTRACT Density Functional methods were applied to systems of chemical interest. First, the effects of integration grid quadrature choice upon energy precision were documented. This was done through application of DFT theory as implemented in five standard computational chemistry programs to a subset of the G2/97 test set of molecules. Subsequently, the neutral hydrogen-loss radicals of naphthalene, anthracene, tetracene, and pentacene and their anions where characterized using five standard DFT treatments. The global and local electron affinities were computed for the twelve radicals. The results for the 1- naphthalenyl and 2-naphthalenyl radicals were compared to experiment, and it was found that B3LYP appears to be the most reliable functional for this type of system. For the larger systems the predicted site specific adiabatic electron affinities of the radicals are 1.51 eV (1-anthracenyl), 1.46 eV (2-anthracenyl), 1.68 eV (9-anthracenyl); 1.61 eV (1-tetracenyl), 1.56 eV (2-tetracenyl), 1.82 eV (12-tetracenyl); 1.93 eV (14-pentacenyl), 2.01 eV (13-pentacenyl), 1.68 eV (1-pentacenyl), and 1.63 eV (2-pentacenyl). The global minimum for each radical does not have the same hydrogen removed as the global minimum for the analogous anion. With this in mind, the global (or most preferred site) adiabatic electron affinities are 1.37 eV (naphthalenyl), 1.64 eV (anthracenyl), 1.81 eV (tetracenyl), and 1.97 eV (pentacenyl). In later work, ten (scandium through zinc) homonuclear transition metal trimers were studied using one DFT 2 functional. -
![Arxiv:1710.00259V1 [Physics.Chem-Ph] 30 Sep 2017 a Relativistically Correct Electron Interaction](https://docslib.b-cdn.net/cover/2561/arxiv-1710-00259v1-physics-chem-ph-30-sep-2017-a-relativistically-correct-electron-interaction-842561.webp)
Arxiv:1710.00259V1 [Physics.Chem-Ph] 30 Sep 2017 a Relativistically Correct Electron Interaction
One-step treatment of spin-orbit coupling and electron correlation in large active spaces Bastien Mussard1, ∗ and Sandeep Sharma1, y 1Department of Chemistry and Biochemistry, University of Colorado Boulder, Boulder, CO 80302, USA In this work we demonstrate that the heat bath configuration interaction (HCI) and its semis- tochastic extension can be used to treat relativistic effects and electron correlation on an equal footing in large active spaces to calculate the low energy spectrum of several systems including halogens group atoms (F, Cl, Br, I), coinage atoms (Cu, Au) and the Neptunyl(VI) dioxide radical. This work demonstrates that despite a significant increase in the size of the Hilbert space due to spin symmetry breaking by the spin-orbit coupling terms, HCI retains the ability to discard large parts of the low importance Hilbert space to deliver converged absolute and relative energies. For instance, by using just over 107 determinants we get converged excitation energies for Au atom in an active space containing (150o,25e) which has over 1030 determinants. We also investigate the accuracy of five different two-component relativistic Hamiltonians in which different levels of ap- proximations are made in deriving the one-electron and two-electrons Hamiltonians, ranging from Breit-Pauli (BP) to various flavors of exact two-component (X2C) theory. The relative accuracy of the different Hamiltonians are compared on systems that range in atomic number from first row atoms to actinides. I. INTRODUCTION can become large and it is more appropriate to treat them on an equal footing with the spin-free terms and electron Relativistic effects play an important role in a correlation. -

Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C
Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C. All rights reserved. Schrödinger, FirstDiscovery, Glide, Impact, Jaguar, Liaison, LigPrep, Maestro, Prime, QSite, and QikProp are trademarks of Schrödinger, L.L.C. MacroModel is a registered trademark of Schrödinger, L.L.C. To the maximum extent permitted by applicable law, this publication is provided “as is” without warranty of any kind. This publication may contain trademarks of other companies. October 2003 Contents Chapter 1: Introduction.......................................................................................1 1.1 Conventions Used in This Manual.......................................................................2 1.2 Citing Jaguar in Publications ...............................................................................3 Chapter 2: The Maestro Graphical User Interface...........................................5 2.1 Starting Maestro...................................................................................................5 2.2 The Maestro Main Window .................................................................................7 2.3 Maestro Projects ..................................................................................................7 2.4 Building a Structure.............................................................................................9 2.5 Atom Selection ..................................................................................................10 2.6 Toolbar Controls ................................................................................................11 -

A Summary of ERCAP Survey of the Users of Top Chemistry Codes
A survey of codes and algorithms used in NERSC chemical science allocations Lin-Wang Wang NERSC System Architecture Team Lawrence Berkeley National Laboratory We have analyzed the codes and their usages in the NERSC allocations in chemical science category. This is done mainly based on the ERCAP NERSC allocation data. While the MPP hours are based on actually ERCAP award for each account, the time spent on each code within an account is estimated based on the user’s estimated partition (if the user provided such estimation), or based on an equal partition among the codes within an account (if the user did not provide the partition estimation). Everything is based on 2007 allocation, before the computer time of Franklin machine is allocated. Besides the ERCAP data analysis, we have also conducted a direct user survey via email for a few most heavily used codes. We have received responses from 10 users. The user survey not only provide us with the code usage for MPP hours, more importantly, it provides us with information on how the users use their codes, e.g., on which machine, on how many processors, and how long are their simulations? We have the following observations based on our analysis. (1) There are 48 accounts under chemistry category. This is only second to the material science category. The total MPP allocation for these 48 accounts is 7.7 million hours. This is about 12% of the 66.7 MPP hours annually available for the whole NERSC facility (not accounting Franklin). The allocation is very tight. The majority of the accounts are only awarded less than half of what they requested for. -

CHEM:4480 Introduction to Molecular Modeling
CHEM:4480:001 Introduction to Molecular Modeling Fall 2018 Instructor Dr. Sara E. Mason Office: W339 CB, Phone: (319) 335-2761, Email: [email protected] Lecture 11:00 AM - 12:15 PM, Tuesday/Thursday. Primary Location: W228 CB. Please note that we will often meet in one of the Chemistry Computer Labs. Notice about class being held outside of the primary location will be posted to the course ICON site by 24 hours prior to the start of class. Office Tuesday 12:30-2 PM (EXCEPT for DoC faculty meeting days 9/4, 10/2, 10/9, 11/6, Hour and 12/4), Thursday 12:30-2, or by appointment Department Dr. James Gloer DEO Administrative Office: E331 CB Administrative Phone: (319) 335-1350 Administrative Email: [email protected] Text Instead of a course text, assigned readings will be given throughout the semester in forms such as journal articles or library reserves. Website http://icon.uiowa.edu Course To develop practical skills associated with chemical modeling based on quantum me- Objectives chanics and computational chemistry such as working with shell commands, math- ematical software, and modeling packages. We will also work to develop a basic understanding of quantum mechanics, approximate methods, and electronic struc- ture. Technical computing, pseudopotential generation, density functional theory software, (along with other optional packages) will be used hands-on to gain experi- ence in data manipulation and modeling. We will use both a traditional classroom setting and hands-on learning in computer labs to achieve course goals. Course Topics • Introduction: It’s a quantum world! • Dirac notation and matrix mechanics (a.k.a. -
![Trends in Atomistic Simulation Software Usage [1.3]](https://docslib.b-cdn.net/cover/7978/trends-in-atomistic-simulation-software-usage-1-3-1207978.webp)
Trends in Atomistic Simulation Software Usage [1.3]
A LiveCoMS Perpetual Review Trends in atomistic simulation software usage [1.3] Leopold Talirz1,2,3*, Luca M. Ghiringhelli4, Berend Smit1,3 1Laboratory of Molecular Simulation (LSMO), Institut des Sciences et Ingenierie Chimiques, Valais, École Polytechnique Fédérale de Lausanne, CH-1951 Sion, Switzerland; 2Theory and Simulation of Materials (THEOS), Faculté des Sciences et Techniques de l’Ingénieur, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland; 3National Centre for Computational Design and Discovery of Novel Materials (MARVEL), École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland; 4The NOMAD Laboratory at the Fritz Haber Institute of the Max Planck Society and Humboldt University, Berlin, Germany This LiveCoMS document is Abstract Driven by the unprecedented computational power available to scientific research, the maintained online on GitHub at https: use of computers in solid-state physics, chemistry and materials science has been on a continuous //github.com/ltalirz/ rise. This review focuses on the software used for the simulation of matter at the atomic scale. We livecoms-atomistic-software; provide a comprehensive overview of major codes in the field, and analyze how citations to these to provide feedback, suggestions, or help codes in the academic literature have evolved since 2010. An interactive version of the underlying improve it, please visit the data set is available at https://atomistic.software. GitHub repository and participate via the issue tracker. This version dated August *For correspondence: 30, 2021 [email protected] (LT) 1 Introduction Gaussian [2], were already released in the 1970s, followed Scientists today have unprecedented access to computa- by force-field codes, such as GROMOS [3], and periodic tional power. -

The Molpro Quantum Chemistry Package
The Molpro Quantum Chemistry package Hans-Joachim Werner,1, a) Peter J. Knowles,2, b) Frederick R. Manby,3, c) Joshua A. Black,1, d) Klaus Doll,1, e) Andreas Heßelmann,1, f) Daniel Kats,4, g) Andreas K¨ohn,1, h) Tatiana Korona,5, i) David A. Kreplin,1, j) Qianli Ma,1, k) Thomas F. Miller, III,6, l) Alexander Mitrushchenkov,7, m) Kirk A. Peterson,8, n) Iakov Polyak,2, o) 1, p) 2, q) Guntram Rauhut, and Marat Sibaev 1)Institut f¨ur Theoretische Chemie, Universit¨at Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany 2)School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, United Kingdom 3)School of Chemistry, University of Bristol, Cantock’s Close, Bristol BS8 1TS, United Kingdom 4)Max-Planck Institute for Solid State Research, Heisenbergstraße 1, 70569 Stuttgart, Germany 5)Faculty of Chemistry, University of Warsaw, L. Pasteura 1 St., 02-093 Warsaw, Poland 6)Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, United States 7)MSME, Univ Gustave Eiffel, UPEC, CNRS, F-77454, Marne-la- Vall´ee, France 8)Washington State University, Department of Chemistry, Pullman, WA 99164-4630 1 Molpro is a general purpose quantum chemistry software package with a long devel- opment history. It was originally focused on accurate wavefunction calculations for small molecules, but now has many additional distinctive capabilities that include, inter alia, local correlation approximations combined with explicit correlation, highly efficient implementations of single-reference correlation methods, robust and efficient multireference methods for large molecules, projection embedding and anharmonic vibrational spectra.