Characterization of the Complete Mitochondrial Genome of Leucoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ARTHROPOD COMMUNITIES and PASSERINE DIET: EFFECTS of SHRUB EXPANSION in WESTERN ALASKA by Molly Tankersley Mcdermott, B.A./B.S
Arthropod communities and passerine diet: effects of shrub expansion in Western Alaska Item Type Thesis Authors McDermott, Molly Tankersley Download date 26/09/2021 06:13:39 Link to Item http://hdl.handle.net/11122/7893 ARTHROPOD COMMUNITIES AND PASSERINE DIET: EFFECTS OF SHRUB EXPANSION IN WESTERN ALASKA By Molly Tankersley McDermott, B.A./B.S. A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science in Biological Sciences University of Alaska Fairbanks August 2017 APPROVED: Pat Doak, Committee Chair Greg Breed, Committee Member Colleen Handel, Committee Member Christa Mulder, Committee Member Kris Hundertmark, Chair Department o f Biology and Wildlife Paul Layer, Dean College o f Natural Science and Mathematics Michael Castellini, Dean of the Graduate School ABSTRACT Across the Arctic, taller woody shrubs, particularly willow (Salix spp.), birch (Betula spp.), and alder (Alnus spp.), have been expanding rapidly onto tundra. Changes in vegetation structure can alter the physical habitat structure, thermal environment, and food available to arthropods, which play an important role in the structure and functioning of Arctic ecosystems. Not only do they provide key ecosystem services such as pollination and nutrient cycling, they are an essential food source for migratory birds. In this study I examined the relationships between the abundance, diversity, and community composition of arthropods and the height and cover of several shrub species across a tundra-shrub gradient in northwestern Alaska. To characterize nestling diet of common passerines that occupy this gradient, I used next-generation sequencing of fecal matter. Willow cover was strongly and consistently associated with abundance and biomass of arthropods and significant shifts in arthropod community composition and diversity. -

November December 2010 Vol 67.3 Victoria Natural
NOVEMBER DECEMBER 2010 VOL 67.3 VICTORIA NATURAL HISTORY SOCIETY The Victoria Naturalist Vol. 67.3 (2010) 1 Published six times a year by the SUBMISSIONS VICTORIA NATURAL HISTORY SOCIETY, P.O. Box 5220, Station B, Victoria, BC V8R 6N4 Deadline for next issue: December 1, 2010 Contents © 2010 as credited. Send to: Claudia Copley ISSN 0049—612X Printed in Canada 657 Beaver Lake Road, Victoria BC V8Z 5N9 Editors: Claudia Copley, 250-479-6622, Penelope Edwards Phone: 250-479-6622 Desktop Publishing: Frances Hunter, 250-479-1956 e-mail: [email protected] Distribution: Tom Gillespie, Phyllis Henderson, Morwyn Marshall Printing: Fotoprint, 250-382-8218 Guidelines for Submissions Opinions expressed by contributors to The Victoria Naturalist Members are encouraged to submit articles, field trip reports, natural are not necessarily those of the Society. history notes, and book reviews with photographs or illustrations if possible. Photographs of natural history are appreciated along with VICTORIA NATURAL HISTORY SOCIETY documentation of location, species names and a date. Please label your Honorary Life Members Dr. Bill Austin, Mrs. Lyndis Davis, submission with your name, address, and phone number and provide a Mr. Tony Embleton, Mr. Tom Gillespie, Mrs. Peggy Goodwill, title. We request submission of typed, double-spaced copy in an IBM compatible word processing file on diskette, or by e-mail. Photos and Mr. David Stirling, Mr. Bruce Whittington slides, and diskettes submitted will be returned if a stamped, self- Officers: 2009-2010 addressed envelope is included with the material. Digital images are PRESIDENT: Darren Copley, 250-479-6622, [email protected] welcome, but they need to be high resolution: a minimum of 1200 x VICE-PRESIDENT: James Miskelly, 250-477-0490, [email protected] 1550 pixels, or 300 dpi at the size of photos in the magazine. -

(12) Patent Application Publication (10) Pub. No.: US 2014/0148493 A1 Tamura Et Al
US 20140148493A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0148493 A1 Tamura et al. (43) Pub. Date: May 29, 2014 (54) AGRICULTURAL AND HORTICULTURAL Publication Classification INSECTICDE COMPOSITION AND METHOD FORUSING THE SAME (51) Int. Cl. AOIN 43/56 (2006.01) (75) Inventors: Shingo Tamura, Tokyo (JP); Takao AOIN4I/O (2006.01) Aoki, Osaka (JP); Ken Kuriyama, (52) U.S. Cl. Tokyo (JP) CPC ................ A0IN 43/56 (2013.01); A0IN 41/10 (2013.01) (73) Assignee: NIHON NOHYAKU CO.,LTD., Tokyo USPC .......................................................... 514/406 (JP) (21) Appl. No.: 14/125,517 (57) ABSTRACT (22) PCT Fled: Jun. 28, 2012 Provided are an agricultural and horticultural insecticide composition comprising flubendiamide and tolfenpyrad as (86) PCT NO.: active ingredients; and a method for using an agricultural and S371 (c)(1), horticultural insecticide composition, comprising treating (2), (4) Date: Feb. 12, 2014 insect pests directly, treating crops potentially infested with the insect pests, or treating Surrounding soil or cultivation (30) Foreign Application Priority Data medium of the crops with an effective amount of an agricul tural and horticultural insecticide composition comprising Jun. 29, 2011 (JP) ................................. 2011-144372 flubendiamide and tolfenpyrad as active ingredients. US 2014/0148493 A1 May 29, 2014 AGRICULTURAL AND HORTICULTURAL control cannot be achieved in some cases because of the kind INSECTICDE COMPOSITION AND of insect pest to be controlled, habitat characteristics, devel METHOD FOR USING THE SAME opment of insecticide resistance, etc. Under Such circum stances, the development of novel technologies for efficiently TECHNICAL FIELD controlling insect pests that have been difficult or impossible 0001. -

International Standard Bible Encyclopedia Vol. 5 H-Izziah
THE AGES DIGITAL LIBRARY REFERENCE INTERNATIONAL STANDARD BIBLE ENCYCLOPEDIA VOL. 5 H-IZZIAH Books For The Ages AGES Software • Albany, OR USA Version 1.0 © 1997 2 H HA <ha> ([ja;h,, he’-ah]): In Job 39:25, the Revised Version (British and American) “Aha,” of the battle-horse. See AH, AHA. HAAHASHTARI <ha-a-hash’-ta-ri> ([yriT;v]j”a}h;, ha’-achashtari], possibly a corruption of [yriWjv]a”h;, ha’-ashchuri]): A descendant of Judah (1 Ch 4:6). The name is probably corrupt. If the emendation suggested above is accepted, it means the Ashurites, and is a description of the preceding names. HABAIAH; HOBAIAH <ha-ba’-ya>, ([hy;b;j}, cha-bhayah], [hy;b;jÜ, chobhayah]): A post-exilic priestly family which was unable to establish its pedigree. “Habaiah” is the form in Ezr 2:61; in the parallel passage (Neh 7:63), the King James Version has “Habaiah,” and the Revised Version (British and American) “Hobaiah”; in the parallel passage in 1 Esdras 5:38, the form is [ jObdia>, Obdia], Codex Vaticanus, Obbeia. HABAKKUK <ha-bak’-uk>, <hab’-a-kuk>: I. THE AUTHOR. 1. Name: Habakkuk ([qWQb”j}, chabhaqquq]) means “embrace,” or “ardent embrace.” Some of the ancient rabbis, connecting the name with 2 Ki 4:16, “Thou shalt embrace a son,” imagined that the prophet was the son of the Shunammite woman. The Septuagint form of the name, Hambakoum; 3 Theodotion Hambakouk, presupposes the Hebrew chabbaquq. A similar word occurs in Assyrian as the name of a garden plant. 2. Life: Practically nothing is known of Habakkuk. -

The Complete Mitochondrial Genome of Spilonota Lechriaspis Meyrick (Lepidoptera: Tortricidae)
Mol Biol Rep (2011) 38:3757–3764 DOI 10.1007/s11033-010-0491-6 The complete mitochondrial genome of Spilonota lechriaspis Meyrick (Lepidoptera: Tortricidae) Jin-Liang Zhao • Yan-Yan Zhang • A-Rong Luo • Guo-Fang Jiang • Stephen L. Cameron • Chao-Dong Zhu Received: 22 July 2010 / Accepted: 9 November 2010 / Published online: 24 November 2010 Ó Springer Science+Business Media B.V. 2010 Abstract We determined the nucleotide sequence of the Introduction mitochondrial genome (mtgenome) of Spilonota lechria- spis Meyrick (Lepidoptera: Tortricidae). The entire closed Complete mitochondrial genome (mtgenome) sequences circular molecule is 15,368 bp and contains 37 genes with are seeing wider use as molecular markers for phyloge- the typical gene complement and order for lepidopteran netic, phylogeographic and ecological studies of insects mtgenomes. All tRNAs except tRNASer(AGN) can be folded [1–5] plus as a model system for genome biology [6–9]. into the typical cloverleaf secondary structures. The pro- Wolstenholme and Clary [10] reported the first insect tein-coding genes (PCGs) have typical mitochondrial start mtgenome sequence in 1985 for Drosophila. Up to 2006, codons, with the exception of COI, which uses the unusual there had been 46 complete or near complete mtgenome CGA one as is found in all other Lepidoptera sequenced to sequences lodged in GenBank [11], and the number has date. In addition, six of 13 PCGs harbor the incomplete rapidly increased in the last five years, with to 198 ones in termination codons, a single T. The A ? T-rich region April, 2010. The mtgenome of insects as with other met- contains some conserved structures that are similar to those azoans is a closed-circular molecule ranging in size found in other lepidopteran mtgenomes, including a from 14 to 20 kb, which encodes 37 genes, consisting structure combining the motif ‘ATAGA’, a 19-bp poly(T) of 13 PCGs, two ribosomal RNA (rRNA) genes, and 22 stretch and three microsatellite (AT)n elements which are transfer RNA (tRNA) genes [12]. -

1996 No. 4 December
TROPICAL LEPIDOPTERA NEWS December 1996 No.4 LEPIDOPTERORUM CATALOGUS (New Series) The new world catalog of Lepidoptera renews the series title The new series (as edited by J. B. Heppner) began already in first begun in 1911. The original catalog series was published by 1989 with publication of the catalog of Noctuidae, by R. Poole. W. Junk Publishers of Berlin, Germany (later The Hague, E. J. Brill Publishers, of Leiden, Netherlands, published this first Netherlands), continuing until 1939 when the incomplete series fascicle in 3 volumes, covering already about a third of all known was deactivated due to World War II. The original series Lepidoptera. Since ATL took over the series, several families completed a large number of families between 1911 and 1939, have been readied for publication. Already this month, Fascicle totalling about 3 shelf-feet of text. Most Microlepidoptera, 48, on Epermeniidae, was published (authored by R. Gaedike, of however, were not covered, as also several macro families like the Deutsches Entomologisches Institut, Eberswalde, Germany). Noctuidae, and several families are incomplete (e.g., Geometridae In 1997, several other smaller families are expected, including and Pyralidae). Even for what was treated, the older catalogs are Acanthopteroctetidae (Davis), Acrolepiidae (Gaedike), Cecidosi now greatly out of date, due to the description of many new dae (Davis), Cercophanidae (Becker), Glyphipterigidae (Heppner), species and many changes in nomenclature over the last 5 to 8 Neotheoridae (Kristensen), Ochsenheimeriidae (Davis), Opostegi decades. dae (Davis), and Oxytenidae (Becker). Much of the publication The new series resembles the old series in some ways but it schedule depends on the cooperation of various specialists who will also have features not found in the old work. -

Chapter 10 • Principles of Conserving the Arctic's Biodiversity
Chapter 10 Principles of Conserving the Arctic’s Biodiversity Lead Author Michael B. Usher Contributing Authors Terry V.Callaghan, Grant Gilchrist, Bill Heal, Glenn P.Juday, Harald Loeng, Magdalena A. K. Muir, Pål Prestrud Contents Summary . .540 10.1. Introduction . .540 10.2. Conservation of arctic ecosystems and species . .543 10.2.1. Marine environments . .544 10.2.2. Freshwater environments . .546 10.2.3. Environments north of the treeline . .548 10.2.4. Boreal forest environments . .551 10.2.5. Human-modified habitats . .554 10.2.6. Conservation of arctic species . .556 10.2.7. Incorporating traditional knowledge . .558 10.2.8. Implications for biodiversity conservation . .559 10.3. Human impacts on the biodiversity of the Arctic . .560 10.3.1. Exploitation of populations . .560 10.3.2. Management of land and water . .562 10.3.3. Pollution . .564 10.3.4. Development pressures . .566 10.4. Effects of climate change on the biodiversity of the Arctic . .567 10.4.1. Changes in distribution ranges . .568 10.4.2. Changes in the extent of arctic habitats . .570 10.4.3. Changes in the abundance of arctic species . .571 10.4.4. Changes in genetic diversity . .572 10.4.5. Effects on migratory species and their management . .574 10.4.6. Effects caused by non-native species and their management .575 10.4.7. Effects on the management of protected areas . .577 10.4.8. Conserving the Arctic’s changing biodiversity . .579 10.5. Managing biodiversity conservation in a changing environment . .579 10.5.1. Documenting the current biodiversity . .580 10.5.2. -

White Satin Moth
Utah Plant Pest UTAH PESTS Diagnostic Laboratory USU Extension Spring 2018 / QUARTERLY Vol. XII NEWSLETTER IN THIS ISSUE Is Biochar Worth it for Utah’s Vegetable Producers? Is Biochar worth it for Utah’s Vegetable Producers? p. 01 USU Extension Biochar Study p. 03 The Mystery of the Notched Leaves p. 04 New Codling Moth Biofix- Setting Model p. 06 Invasive Pest Spotlight: White Biochar is made by burning biomass at very high temperatures with low oxygen. The final Satin Moth p. 08 product is high in carbon and is used as a soil amendment. Spider Mites Like it Hot with Biochar has been shown to have long- What are the Potential Neonics term benefits for the environment, in terms Advantages in Agriculture? p. 09 of sequestering carbon in the soil. But for 5 Vegetable Insect Pests of intensive agricultural production, the short Most biochars have low nitrogen Utah and When to Watch answer to this question is that biochar is concentrations and thus any inherent for Them not quite ready for wide-scale adoption. fertilizer value is minimal and temporary. p. 11 Some factors that need to be considered in The recommendation is that biochar is Worker Protection Standard applying biochar to the soil are initial soil applied to the soil just once, acting as a Training Requirements health, the source and production method conditioner, and that nutrient amendments for Growers of the char, and the variable or unknown should also be applied yearly (if needed). p. 12 application rates. In some cases, crop yield Some research studies have found the may be marginally increased, but this benefit following benefits of a one-time biochar IPM in the News p. -

Molecular Phylogeny of the Genus Satarupa Moore
國立臺灣師範大學生命科學系碩士論文 颯弄蝶屬之分子親緣與系統研究 Molecular phylogeny and systematics of the genus Satarupa Moore, 1865 (Lepidoptera: Hesperiidae: Pyrginae) 研究生: 莊懷淳 Huai-chun Chuang 指導教授: 徐堉峰 博士 Yu-Feng Hsu 千葉秀幸 博士 Hideyuki Chiba 中華民國 102 年 7 月 致謝 首先要感謝我的父母,經過多番的波折與衝突答應讓我長久以來的心 願能夠一邊實驗一邊做田野調查。並且非常的感謝我的論文指導教 授,徐堉峰教授,給予我如此自由的環境讓我實驗。並且也要感謝實 驗室的各位夥伴,讓整個實驗室充滿了活潑的環境讓原本枯燥的研究 過程增添豐富的色彩,猶如雨天過後的七彩彩虹。感謝實驗室的學長 姐們帶領我野外調查並且獲得台灣難以獲得的樣本。並且也要感謝提 供我國外樣本的千葉秀幸博士,讓我獲得許多困難獲取的樣本,以及 提供我很多論文寫作的相關意見。當然一定要感謝提供如此高規格的 實驗環境的李壽先老師,並且在很多思考上給予很大的啟發,並且教 導我在很多表達上的細節與邏輯。感謝林思民老師在口試與報告的時 候給予我許多中肯的意見。也要感謝共同陪我奮鬥的同學們,一起在 實驗室熬夜趕 ppt 等口頭報告。最後最後一定要感謝這兩年來一起共 同奮鬥的顏嘉瑩同學,在這兩年一起度過了非常多的挫折與試煉,如 果沒有他我這兩年的色彩會黯淡許多。要感謝的人太多,寥寥字詞無 法述說滿滿的感謝,就如陳之藩所言:「要感謝的人太多,不如謝天 吧!」 Contents 中文摘要…………………………...……………………..……1 Abstract…………………………………………………..……2 Introduction………………………………….………….….…4 Materials and methods…………………………………….…8 Results……………………………………………………..….12 Discussion……………………………………………….……14 References...………………………………………………..…18 Tables………………………………………………….……...25 Figures………………………………………………….…….29 Appendix……………………………….……………………37 中文摘要 親緣關係為生物研究的基礎,弄蝶親緣關係已有高階親緣關係發表, 在屬級的親緣關係尚有不足之處。颯弄蝶屬屬特徵為前翅 2A 至 R3 脈之間有透明的斑紋與後翅具有大片的白塊。因颯弄蝶種之間的形態 太過於相似,許多發表尚有辨識錯誤的情形。Evans 在 1949 整理為 7 種,Okano (1987) 和 Chiba (1989) 則認為颯弄蝶屬為 valentini, zulla, gopala, nymphalis, splendens, monbeigi 和 formosibia。以翅形與幼蟲型 態 S. formosibia 與 S. monbeigi 應屬於同一類群,但是前者外生殖器卻 並不典型。Shirôzu 將 S. majasra 處理為 gopala 的亞種,但 Tsukiyama 處理為 nymphalis 的亞種。颯弄蝶屬近緣屬皆為熱帶分布,颯弄蝶卻 分布自爪哇至溫帶的西伯利亞。建構颯弄蝶屬親緣關係可以確立 formosibia 及 majasra 的分類地位並且了解颯弄蝶屬的溫帶分布是由 熱帶分布至溫帶或原本分布在溫帶。本研究使用 COI, COII 與 Ef1α -
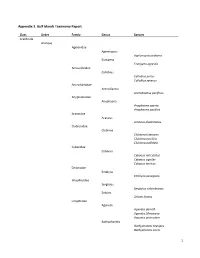
1 Appendix 3. Gulf Islands Taxonomy Report
Appendix 3. Gulf Islands Taxonomy Report Class Order Family Genus Species Arachnida Araneae Agelenidae Agelenopsis Agelenopsis utahana Eratigena Eratigena agrestis Amaurobiidae Callobius Callobius pictus Callobius severus Antrodiaetidae Antrodiaetus Antrodiaetus pacificus Anyphaenidae Anyphaena Anyphaena aperta Anyphaena pacifica Araneidae Araneus Araneus diadematus Clubionidae Clubiona Clubiona lutescens Clubiona pacifica Clubiona pallidula Cybaeidae Cybaeus Cybaeus reticulatus Cybaeus signifer Cybaeus tetricus Dictynidae Emblyna Emblyna peragrata Gnaphosidae Sergiolus Sergiolus columbianus Zelotes Zelotes fratris Linyphiidae Agyneta Agyneta darrelli Agyneta fillmorana Agyneta protrudens Bathyphantes Bathyphantes brevipes Bathyphantes keeni 1 Centromerita Centromerita bicolor Ceratinops Ceratinops latus Entelecara Entelecara acuminata Erigone Erigone aletris Erigone arctica Erigone cristatopalpus Frederickus Frederickus coylei Grammonota Grammonota kincaidi Linyphantes Linyphantes nehalem Linyphantes nigrescens Linyphantes pacificus Linyphantes pualla Linyphantes victoria Mermessus Mermessus trilobatus Microlinyphia Microlinyphia dana Neriene Neriene digna Neriene litigiosa Oedothorax Oedothorax alascensis Pityohyphantes Pityohyphantes alticeps Pocadicnemis Pocadicnemis pumila Poeciloneta Poeciloneta fructuosa Saaristoa Saaristoa sammamish Scotinotylus Scotinotylus sp. 5GAB Semljicola Semljicola sp. 1GAB Sisicottus Spirembolus Spirembolus abnormis Spirembolus mundus Tachygyna Tachygyna ursina Tachygyna vancouverana Tapinocyba Tapinocyba -

Lepidoptera: Noctuoidea: Erebidae) and Its Phylogenetic Implications
EUROPEAN JOURNAL OF ENTOMOLOGYENTOMOLOGY ISSN (online): 1802-8829 Eur. J. Entomol. 113: 558–570, 2016 http://www.eje.cz doi: 10.14411/eje.2016.076 ORIGINAL ARTICLE Characterization of the complete mitochondrial genome of Spilarctia robusta (Lepidoptera: Noctuoidea: Erebidae) and its phylogenetic implications YU SUN, SEN TIAN, CEN QIAN, YU-XUAN SUN, MUHAMMAD N. ABBAS, SAIMA KAUSAR, LEI WANG, GUOQING WEI, BAO-JIAN ZHU * and CHAO-LIANG LIU * College of Life Sciences, Anhui Agricultural University, 130 Changjiang West Road, Hefei, 230036, China; e-mails: [email protected] (Y. Sun), [email protected] (S. Tian), [email protected] (C. Qian), [email protected] (Y.-X. Sun), [email protected] (M.-N. Abbas), [email protected] (S. Kausar), [email protected] (L. Wang), [email protected] (G.-Q. Wei), [email protected] (B.-J. Zhu), [email protected] (C.-L. Liu) Key words. Lepidoptera, Noctuoidea, Erebidae, Spilarctia robusta, phylogenetic analyses, mitogenome, evolution, gene rearrangement Abstract. The complete mitochondrial genome (mitogenome) of Spilarctia robusta (Lepidoptera: Noctuoidea: Erebidae) was se- quenced and analyzed. The circular mitogenome is made up of 15,447 base pairs (bp). It contains a set of 37 genes, with the gene complement and order similar to that of other lepidopterans. The 12 protein coding genes (PCGs) have a typical mitochondrial start codon (ATN codons), whereas cytochrome c oxidase subunit 1 (cox1) gene utilizes unusually the CAG codon as documented for other lepidopteran mitogenomes. Four of the 13 PCGs have incomplete termination codons, the cox1, nad4 and nad6 with a single T, but cox2 has TA. It comprises six major intergenic spacers, with the exception of the A+T-rich region, spanning at least 10 bp in the mitogenome. -

EUROPEAN JOURNAL of ENTOMOLOGYENTOMOLOGY ISSN (Online): 1802-8829 Eur
EUROPEAN JOURNAL OF ENTOMOLOGYENTOMOLOGY ISSN (online): 1802-8829 Eur. J. Entomol. 113: 482–488, 2016 http://www.eje.cz doi: 10.14411/eje.2016.063 ORIGINAL ARTICLE The mitochondrial genome of the Mediterranean fl our moth, Ephestia kuehniella (Lepidoptera: Pyralidae), and identifi cation of invading mitochondrial sequences (numts) in the W chromosome KATRIN LÄMMERMANN 1, 3, HEIKO VOGEL 2 and WALTHER TRAUT 3, * 1 Universität zu Lübeck, Institut für Neuro- und Bioinformatik, Ratzeburger Allee 160, D-23538 Lübeck, Germany; e-mail: [email protected] 2 Max Planck Institute for Chemical Ecology, Department of Entomology, Hans-Knoell-Strasse 8, D-07745 Jena, Germany; e-mail: [email protected] 3 Universität zu Lübeck, Zentrum für Medizinische Strukturbiologie, Institut für Biologie, Ratzeburger Allee 160, D-23538 Lübeck, Germany; e-mail: [email protected] Key words. Lepidoptera, Pyralidae, Ephestia kuehniella, mitogenome, Mediterranean fl our moth, phylogeny, numts, W chromosome Abstract. The Mediterranean fl our moth, Ephestia kuehniella is a widespread pest of stored products and a classical object in experimental biology. In the present study, we determined its complete mitochondrial genome sequence. The genome is circular, consists of 15,327 bp and comprises 13 protein-coding, 2 rRNA- and 22 tRNA-coding genes in an order typical for the Ditrysia clade of the order Lepidoptera. A phylogenetic study of the Lepidoptera based on complete mitochondrial genomes places E. kuehniella correctly in the family Pyralidae and supports major lepidopteran taxa as phylogenetic clades. The W chromosome of E. kuehniella is an exceptionally rich reservoir of originally mitochondrial sequences (numts). Around 0.7% of the W DNA was found to be of mitochondrial origin, 83% of the mitogenome sequence was represented between 1–11 × in the W chromosome.