Neuroprotection by Chlorpromazine and Promethazine in Severe Transient and Permanent Ischemic Stroke
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
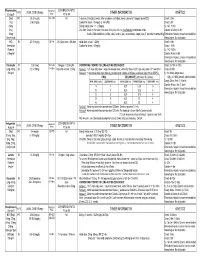
Medication Conversion Chart
Fluphenazine FREQUENCY CONVERSION RATIO ROUTE USUAL DOSE (Range) (Range) OTHER INFORMATION KINETICS Prolixin® PO to IM Oral PO 2.5-20 mg/dy QD - QID NA ↑ dose by 2.5mg/dy Q week. After symptoms controlled, slowly ↓ dose to 1-5mg/dy (dosed QD) Onset: ≤ 1hr 1mg (2-60 mg/dy) Caution for doses > 20mg/dy (↑ risk EPS) Cmax: 0.5hr 2.5mg Elderly: Initial dose = 1 - 2.5mg/dy t½: 14.7-15.3hr 5mg Oral Soln: Dilute in 2oz water, tomato or fruit juice, milk, or uncaffeinated carbonated drinks Duration of Action: 6-8hr 10mg Avoid caffeinated drinks (coffee, cola), tannics (tea), or pectinates (apple juice) 2° possible incompatibilityElimination: Hepatic to inactive metabolites 5mg/ml soln Hemodialysis: Not dialyzable HCl IM 2.5-10 mg/dy Q6-8 hr 1/3-1/2 po dose = IM dose Initial dose (usual): 1.25mg Onset: ≤ 1hr Immediate Caution for doses > 10mg/dy Cmax: 1.5-2hr Release t½: 14.7-15.3hr 2.5mg/ml Duration Action: 6-8hr Elimination: Hepatic to inactive metabolites Hemodialysis: Not dialyzable Decanoate IM 12.5-50mg Q2-3 wks 10mg po = 12.5mg IM CONVERTING FROM PO TO LONG-ACTING DECANOATE: Onset: 24-72hr (4-72hr) Long-Acting SC (12.5-100mg) (1-4 wks) Round to nearest 12.5mg Method 1: 1.25 X po daily dose = equiv decanoate dose; admin Q2-3wks. Cont ½ po daily dose X 1st few mths Cmax: 48-96hr 25mg/ml Method 2: ↑ decanoate dose over 4wks & ↓ po dose over 4-8wks as follows (accelerate taper for sx of EPS): t½: 6.8-9.6dy (single dose) ORAL DECANOATE (Administer Q 2 weeks) 15dy (14-100dy chronic administration) ORAL DOSE (mg/dy) ↓ DOSE OVER (wks) INITIAL DOSE (mg) TARGET DOSE (mg) DOSE OVER (wks) Steady State: 2mth (1.5-3mth) 5 4 6.25 6.25 0 Duration Action: 2wk (1-6wk) Elimination: Hepatic to inactive metabolites 10 4 6.25 12.5 4 Hemodialysis: Not dialyzable 20 8 6.25 12.5 4 30 8 6.25 25 4 40 8 6.25 25 4 Method 3: Admin equivalent decanoate dose Q2-3wks. -

Is Aristada (Aripiprazole Lauroxil) a Safe and Effective Treatment for Schizophrenia in Adult Patients? Kyle J
Philadelphia College of Osteopathic Medicine DigitalCommons@PCOM PCOM Physician Assistant Studies Student Student Dissertations, Theses and Papers Scholarship 2017 Is Aristada (Aripiprazole Lauroxil) a Safe and Effective Treatment For Schizophrenia In Adult Patients? Kyle J. Knowles Philadelphia College of Osteopathic Medicine Follow this and additional works at: https://digitalcommons.pcom.edu/pa_systematic_reviews Part of the Psychiatry Commons Recommended Citation Knowles, Kyle J., "Is Aristada (Aripiprazole Lauroxil) a Safe and Effective Treatment For Schizophrenia In Adult Patients?" (2017). PCOM Physician Assistant Studies Student Scholarship. 381. https://digitalcommons.pcom.edu/pa_systematic_reviews/381 This Selective Evidence-Based Medicine Review is brought to you for free and open access by the Student Dissertations, Theses and Papers at DigitalCommons@PCOM. It has been accepted for inclusion in PCOM Physician Assistant Studies Student Scholarship by an authorized administrator of DigitalCommons@PCOM. For more information, please contact [email protected]. Is Aristada (Aripiprazole Lauroxil) a Safe and Effective Treatment For Schizophrenia In Adult Patients? Kyle J. Knowles, PA-S A SELECTIVE EVIDENCE BASED MEDICINE REVIEW In Partial Fulfillment of the Requirements For The Degree of Master of Science In Health Sciences- Physician Assistant Department of Physician Assistant Studies Philadelphia College of Osteopathic Medicine Philadelphia, Pennsylvania December 16, 2016 ABSTRACT OBJECTIVE: The objective of this selective EBM review is to determine whether or not “Is Aristada (aripiprazole lauroxil) a safe and effective treatment for schizophrenia in adult patients?” STUDY DESIGN: Review of three randomized controlled studies. All three trials were conducted between 2014 and 2015. DATA SOURCES: One randomized, controlled trial and two randomized, controlled, double- blind trials found via Cochrane Library and PubMed. -

Antipsychotic Combinations
Graylands Hospital DRUG BULLETIN Pharmacy Department Brockway Road Mount Claremont WA 6010 Telephone (08) 9347 6400 Email [email protected] Fax (08) 9384 4586 Antipsychotic Combinations Graylands Hospital Drug Bulletin 2008 Vol. 15 No. 3 February ISSN 1323-1251 Antipsychotic combinations Reasons for antipsychotic combinations Despite the development of efficacious medications for the treatment of schizophrenia, many people do There are a number of theoretical benefits and not respond adequately. To address this problem, reasons cited for antipsychotic combination the use of two or more antipsychotics simultaneously prescribing, these include: is a commonly employed treatment strategy. Although the use of combination antipsychotics is Complementary mechanisms of action4 (e.g. common in clinical practice, the risks and benefits adding an antipsychotic with strong dopamine have not been systematically evaluated to date. As a D2 blockade to a weak D2 blocker) result, current Australian treatment algorithms Partial replacement of antipsychotic action for including the Royal Australian and New Zealand drugs with intolerable adverse effects at higher College of Psychiatry Schizophrenia Guidelines and 5 doses (e.g. adding quetiapine to clozapine to the Western Australian Therapeutic Advisory Group minimise metabolic adverse effects) Antipsychotic Guidelines advise against the use of combined antipsychotics, except for short periods of Alternative where clozapine cannot be used6 changeover1,2. Most data on antipsychotic -

New Drugs Are Not Enough‑Drug Repositioning in Oncology: an Update
INTERNATIONAL JOURNAL OF ONCOLOGY 56: 651-684, 2020 New drugs are not enough‑drug repositioning in oncology: An update ROMINA GABRIELA ARMANDO, DIEGO LUIS MENGUAL GÓMEZ and DANIEL EDUARDO GOMEZ Laboratory of Molecular Oncology, Science and Technology Department, National University of Quilmes, Bernal B1876, Argentina Received August 15, 2019; Accepted December 16, 2019 DOI: 10.3892/ijo.2020.4966 Abstract. Drug repositioning refers to the concept of discov- 17. Lithium ering novel clinical benefits of drugs that are already known 18. Metformin for use treating other diseases. The advantages of this are that 19. Niclosamide several important drug characteristics are already established 20. Nitroxoline (including efficacy, pharmacokinetics, pharmacodynamics and 21. Nonsteroidal anti‑inflammatory drugs toxicity), making the process of research for a putative drug 22. Phosphodiesterase-5 inhibitors quicker and less costly. Drug repositioning in oncology has 23. Pimozide received extensive focus. The present review summarizes the 24. Propranolol most prominent examples of drug repositioning for the treat- 25. Riluzole ment of cancer, taking into consideration their primary use, 26. Statins proposed anticancer mechanisms and current development 27. Thalidomide status. 28. Valproic acid 29. Verapamil 30. Zidovudine Contents 31. Concluding remarks 1. Introduction 2. Artesunate 1. Introduction 3. Auranofin 4. Benzimidazole derivatives In previous decades, a considerable amount of work has been 5. Chloroquine conducted in search of novel oncological therapies; however, 6. Chlorpromazine cancer remains one of the leading causes of death globally. 7. Clomipramine The creation of novel drugs requires large volumes of capital, 8. Desmopressin alongside extensive experimentation and testing, comprising 9. Digoxin the pioneer identification of identifiable targets and corrobora- 10. -

PERPHENAZINE and AMITRIPTYLINE HYDROCHLORIDE- Perphenazine and Amitriptyline Hydrochloride Tablet, Film Coated Mylan Pharmaceuticals Inc
PERPHENAZINE AND AMITRIPTYLINE HYDROCHLORIDE- perphenazine and amitriptyline hydrochloride tablet, film coated Mylan Pharmaceuticals Inc. ---------- WARNING Increased Mortality in Elderly Patients with Dementia-Related Psychosis Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of seventeen placebo-controlled trials (modal duration of 10 weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in drug-treated patients of between 1.6 to 1.7 times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug- treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group. Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. Observational studies suggest that, similar to atypical antipsychotic drugs, treatment with conventional antipsychotic drugs may increase mortality. The extent to which the findings of increased mortality in observational studies may be attributed to the antipsychotic drug as opposed to some characteristic(s) of the patients is not clear. Perphenazine and amitriptyline hydrochloride is not approved for the treatment of patients with dementia- related psychosis (see WARNINGS). Suicidality and Antidepressant Drugs Antidepressants increased the risk compared to placebo of suicidal thinking and behavior (suicidality) in children, adolescents and young adults in short-term studies of major depressive disorder (MDD) and other psychiatric disorders. Anyone considering the use of perphenazine and amitriptyline or any other antidepressant in a child, adolescent, or young adult must balance this risk with the clinical need. -

Antidepressant Drugs in Oral Fluid Using Liquid Chromatography –Tandem Mass Spectrometry
Journal of Analytical Toxicology, Vol. 34, March 2010 Antidepressant Drugs in Oral Fluid Using Liquid Chromatography –Tandem Mass Spectrometry Cynthia Coulter, Margaux Taruc, James Tuyay, and Christine Moore* Immunalysis Corporation, 829 Towne Center Drive, Pomona, California 91767 Abstract graines, or sleep disorders. Antidepressant drugs may also cause impairment of cognitive and psychomotor functioning An analytical procedure for the determination of widely relevant to driving, although this impairment is reportedly prescribed drugs for the treatment of depression and anxiety less severe in second generation antidepressants such as se - disorders, including amitriptyline, cyclobenzaprine, imipramine, lective serotonin reuptake inhibitors (SSRIs) than with more dothiepin, doxepin, fluoxetine, sertraline, trimipramine, traditional tricyclic antidepressants (TCAs) such as amitripty - protriptyline, chlorpromazine, clomipramine, and some of their line (1,2). Patients receiving long-term treatment with SSRI metabolites (nortriptyline, desmethyldoxepin, desipramine, and serotonin-norepinephrine reuptake inhibitor (SNRI)-type desmethyltrimipramine, norclomipramine) in oral fluid has been drugs produce impaired driving performance (3). However, developed and validated using liquid chromatography with tandem Brunnauer et al. (2) showed that there were actually no sta - mass spectral detection. The oral fluid samples were collected using the Quantisal™ device, screened with enzyme-linked tistically significant differences between patients treated -

Promethazine-Chlorpromazine Combination in the Treatment of Unmanageable Psychotic Patients Armando R
Henry Ford Hospital Medical Journal Volume 17 | Number 4 Article 10 12-1969 Promethazine-Chlorpromazine Combination in the Treatment of Unmanageable Psychotic Patients Armando R. Favazza Follow this and additional works at: https://scholarlycommons.henryford.com/hfhmedjournal Part of the Chemicals and Drugs Commons, Life Sciences Commons, Medical Specialties Commons, Psychiatry and Psychology Commons, and the Public Health Commons Recommended Citation Favazza, Armando R. (1969) "Promethazine-Chlorpromazine Combination in the Treatment of Unmanageable Psychotic Patients," Henry Ford Hospital Medical Journal : Vol. 17 : No. 4 , 305-310. Available at: https://scholarlycommons.henryford.com/hfhmedjournal/vol17/iss4/10 This Article is brought to you for free and open access by Henry Ford Health System Scholarly Commons. It has been accepted for inclusion in Henry Ford Hospital Medical Journal by an authorized editor of Henry Ford Health System Scholarly Commons. Henry Ford Hosp. Med. Journal VoL 17, No. 4, 1969 Promethazine-chlorpromazine Combination in the Treatment of Unmanageable Psychotic Patients Armando R. Favazza, M.D.* Administering a combination of promethazine and chlorpromazine to patients with a "galloping psychosis" has an antipsychotic and tranquilizing effect which calms them down to a more manageable and less aggressive state. The drugs are chemically similar; promethazine's actions are strongly potentiated in the combina tion, so large doses must be given under careful supervision. Case histories demonstrate successful short term management of acutely psychotic, aggressive patients who were a danger to themselves and others. Acute, intense psychotic episodes common in the author's experience for can pose difficult problems for both some patients to receive massive doses the patient and those who care for him. -

Name of Medicine
HALDOL® haloperidol decanoate NEW ZEALAND DATA SHEET 1. PRODUCT NAME HALDOL haloperidol decanoate 50 mg/mL Injection HALDOL CONCENTRATE haloperidol decanoate 100 mg/mL Injection 2. QUANTITATIVE AND QUALITATIVE COMPOSITION HALDOL 50 mg/ml Haloperidol decanoate 70.52 mg, equivalent to 50 mg haloperidol base, per millilitre. HALDOL CONCENTRATE 100 mg/ml Haloperidol decanoate 141.04 mg, equivalent to 100 mg haloperidol base, per millilitre. For a full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Injection (depot) HALDOL Injection (long acting) is a slightly amber, slightly viscous solution, free from visible foreign matter, filled in 1 mL amber glass ampoules. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications HALDOL is indicated for the maintenance therapy of psychoses in adults, particularly for patients requiring prolonged parenteral neuroleptic therapy. 4.2 Dose and method of administration Administration HALDOL should be administered by deep intramuscular injection into the gluteal region. It is recommended to alternate between the two gluteal muscles for subsequent injections. A 2 inch-long, 21 gauge needle is recommended. The maximum volume per injection site should not exceed 3 mL. The recommended interval between doses is 4 weeks. DO NOT ADMINISTER INTRAVENOUSLY. Patients must be previously stabilised on oral haloperidol before converting to HALDOL. Treatment initiation and dose titration must be carried out under close clinical supervision. The starting dose of HALDOL should be based on the patient's clinical history, severity of symptoms, physical condition and response to the current oral haloperidol dose. Patients must always be maintained on the lowest effective dose. CCDS(180302) Page 1 of 16 HALDOL(180712)ADS Dosage - Adults Table 1. -

207533Orig1s000
( CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 207533Orig1s000 RISK ASSESSMENT and RISK MITIGATION REVIEW(S) Department of Health and Human Services Public Health Service Food and Drug Administration Center for Drug Evaluation and Research Office of Surveillance and Epidemiology Office of Medication Error Prevention and Risk Management Risk Evaluation and Mitigation Strategy (REMS) Review Date: October 2, 2015 Reviewer(s): Cathy A. Miller, MPH, B.SN Risk Management Analyst Division of Risk Management Team Leader Kimberly Lehrfeld, Pharm.D. Division of Risk Management Acting Deputy Division Reema Mehta, Pharm.D., MPH Director Division of Risk Management Drug Name(s): Aristada (aripiprazole lauroxil) extended-release injection Therapeutic Class: Atypical Antipsychotic Dosage and Route: 441 mg/1.6 mL; 662 mg/2.4 mL; 882 mg/3.2mL intramuscular injection Application Type/Number: NDA 207533 Submission Number: ORIG-1 Submission Seq. No. 0000 dated August 22, 2014 Applicant/Sponsor: Alkermes OSE RCM #: 2014-1849 ***This document contains proprietary and confidential information that should not be released to the public*** Reference ID: 3828483 CONTENTS 1 INTRODUCTION ....................................................................................................... 1 1.1 Product Background............................................................................................ 1 1.2 Disease Background............................................................................................ 2 1.3 Regulatory History ............................................................................................. -

Chlorpromazine Versus Atypical Antipsychotic Drugs for Schizophrenia (Review)
Cochrane Database of Systematic Reviews Chlorpromazine versus atypical antipsychotic drugs for schizophrenia (Review) Saha KB, Bo L, Zhao S, Xia J, Sampson S, Zaman RU SahaKB, Bo L, ZhaoS, Xia J, Sampson S,ZamanRU. Chlorpromazine versus atypical antipsychotic drugs for schizophrenia. Cochrane Database of Systematic Reviews 2016, Issue 4. Art. No.: CD010631. DOI: 10.1002/14651858.CD010631.pub2. www.cochranelibrary.com Chlorpromazine versus atypical antipsychotic drugs for schizophrenia (Review) Copyright © 2016 The Cochrane Collaboration. Published by John Wiley & Sons, Ltd. TABLE OF CONTENTS HEADER....................................... 1 ABSTRACT ...................................... 1 PLAINLANGUAGESUMMARY . 2 SUMMARY OF FINDINGS FOR THE MAIN COMPARISON . ..... 4 BACKGROUND .................................... 8 OBJECTIVES ..................................... 9 METHODS ...................................... 9 RESULTS....................................... 15 Figure1. ..................................... 17 Figure2. ..................................... 24 Figure3. ..................................... 25 Figure4. ..................................... 27 ADDITIONALSUMMARYOFFINDINGS . 45 DISCUSSION ..................................... 52 AUTHORS’CONCLUSIONS . 54 ACKNOWLEDGEMENTS . 55 REFERENCES ..................................... 55 CHARACTERISTICSOFSTUDIES . 65 DATAANDANALYSES. 150 Analysis 1.1. Comparison 1 CHLORPROMAZINE versus OLANZAPINE, Outcome 1 Clinical response: 1. No significant clinical response. 161 Analysis 1.2. Comparison -

207533Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 207533Orig1s000 MEDICAL REVIEW(S) CLINICAL REVIEW Application Type NDA Application Number(s) 207553 Priority or Standard Standard Submit Date(s) 8/22/2014 Received Date(s) 8/22/2014 PDUFA Goal Date 8/22/2015 Division / Office DPP/ODE1 Reviewer Name(s) Lucas Kempf, MD Review Completion Date October 2, 2015 Established Name Aripiprazole lauroxil (Proposed) Trade Name Aristada Therapeutic Class Antipsychotic Applicant Alkermes Inc. Formulation(s) Extended release injection Dosing Regimen Suspension/ IM up to 6 weeks Indication(s) Schizophrenia Intended Population(s) Schizophrenia Template Version: March 6, 2009 Reference ID: 3806701 Clinical Review Lucas Kempf, MD NDA 207533 Aristada, Aripiprazole lauroxil Table of Contents 1 RECOMMENDATIONS/RISK BENEFIT ASSESSMENT ......................................... 7 1.1 Recommendation on Regulatory Action ............................................................. 7 1.2 Risk Benefit Assessment .................................................................................... 7 1.3 Recommendations for Postmarket Risk Evaluation and Mitigation Strategies ... 8 1.4 Recommendations for Postmarket Requirements and Commitments ................ 8 2 INTRODUCTION AND REGULATORY BACKGROUND ........................................ 8 2.1 Product Information ............................................................................................ 8 2.2 Currently Available Treatments for Proposed Indications .................................. -

Tricyclic Antidepressant Overdose
Tricyclic Antidepressant Overdose R. Kevin Smith, DO, and Karen O'Mara, DO Chicago, Illinois Overdose from tricyclic antidepressants (TCAs) is increasing. TCAs are well absorbed orally, highly protein bound, and high ly lipid soluble. Clinical features of poisoning with TCAs occur within 12 hours of ingestion, usually after a dose of 20 mg/kg or more. Clinical symptomatology involves various anticholiner gic, central nervous system, and cardiovascular effects. Car diovascular toxicity accounts for the majority of the fatalities and may include a hyperdynamic response, various arrhyth mias and heart blocks, or severe hypotension. Prolongation of the QRS interval of 10 msec or more implies severe toxicity. Many factors limit the usefulness of drug levels in the over dosed patient. Treatment revolves around good supportive care and general poisoning management. The physician should no longer use physostigmine precipitously. Sodium bicarbon ate is effective in treating many of the cardiovascular compli cations. Other cardiac drugs are used but with varying effi cacy. Patients with significant signs or symptoms of toxicity require monitored hospitalization until clinically free of mani festations for 24 to 48 hours. Overdosage with tricyclic antidepressant drugs sion, coma with respiratory arrest, convulsions, has been reported as early as 1959, one year after complex arrhythmias, and myocardial depression. their introduction to psychiatry. Because of the The purpose of this paper is to review the current complex central and peripheral toxic effects of body of knowledge on tricyclic antidepressants these drugs, the physician who treats the over (TCAs) and their toxicity. Increased understand dosed patient may find himself managing hypoten- ing of the mechanisms of toxicity, pharmacologic interactions, and clinical manifestations allows the outline of a rational mode of therapy for patients who present with TCA overdose.