Mitochondrial Biogenesis and Dynamics in the Developing and Diseased Heart
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
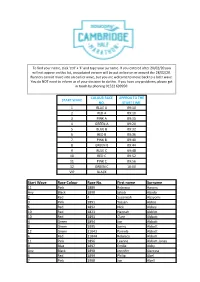
Start Wave Race Colour Race No. First Name Surname
To find your name, click 'ctrl' + 'F' and type your surname. If you entered after 20/02/20 you will not appear on this list, an updated version will be put online on or around the 28/02/20. Runners cannot move into an earlier wave, but you are welcome to move back to a later wave. You do NOT need to inform us of your decision to do this. If you have any problems, please get in touch by phoning 01522 699950. COLOUR RACE APPROX TO THE START WAVE NO. START TIME 1 BLUE A 09:10 2 RED A 09:10 3 PINK A 09:15 4 GREEN A 09:20 5 BLUE B 09:32 6 RED B 09:36 7 PINK B 09:40 8 GREEN B 09:44 9 BLUE C 09:48 10 RED C 09:52 11 PINK C 09:56 12 GREEN C 10:00 VIP BLACK Start Wave Race Colour Race No. First name Surname 11 Pink 1889 Rebecca Aarons Any Black 1890 Jakob Abada 2 Red 4 Susannah Abayomi 3 Pink 1891 Yassen Abbas 6 Red 1892 Nick Abbey 10 Red 1823 Hannah Abblitt 10 Red 1893 Clare Abbott 4 Green 1894 Jon Abbott 8 Green 1895 Jonny Abbott 12 Green 11043 Pamela Abbott 6 Red 11044 Rebecca Abbott 11 Pink 1896 Leanne Abbott-Jones 9 Blue 1897 Emilie Abby Any Black 1898 Jennifer Abecina 6 Red 1899 Philip Abel 7 Pink 1900 Jon Abell 10 Red 600 Kirsty Aberdein 6 Red 11045 Andrew Abery Any Black 1901 Erwann ABIVEN 11 Pink 1902 marie joan ablat 8 Green 1903 Teresa Ablewhite 9 Blue 1904 Ahid Abood 6 Red 1905 Alvin Abraham 9 Blue 1906 Deborah Abraham 6 Red 1907 Sophie Abraham 1 Blue 11046 Mitchell Abrams 4 Green 1908 David Abreu 11 Pink 11047 Kathleen Abuda 10 Red 11048 Annalisa Accascina 4 Green 1909 Luis Acedo 10 Red 11049 Vikas Acharya 11 Pink 11050 Catriona Ackermann -

TRANSCRIPTIONAL REGULATION of Hur in RENAL STRESS
TRANSCRIPTIONAL REGULATION OF HuR IN RENAL STRESS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Sudha Suman Govindaraju Graduate Program in Biochemistry The Ohio State University 2014 Dissertation Committee: Dr. Beth S. Lee, Ph.D., Advisor Dr. Kathleen Boris-Lawrie, Ph.D. Dr. Sissy M. Jhiang, Ph.D. Dr. Arthur R. Strauch, Ph.D Abstract HuR is a ubiquitously expressed RNA-binding protein that affects the post- transcriptional life of thousands of cellular mRNAs by regulating transcript stability and translation. HuR can post-transcriptionally regulate gene expression and modulate cellular responses to stress, differentiation, proliferation, apoptosis, senescence, inflammation, and the immune response. It is an important mediator of survival during cellular stress, but when inappropriately expressed, can promote oncogenic transformation. Not surprisingly, the expression of HuR itself is tightly regulated at multiple transcriptional and post-transcriptional levels. Previous studies demonstrated the existence of two alternate HuR transcripts that differ in their 5’ untranslated regions and have markedly different translatabilities. These forms were also found to be reciprocally expressed following cellular stress in kidney proximal tubule cell lines, and the shorter, more readily translatable variant was shown to be regulated by Smad 1/5/8 pathway and bone morphogenetic protein-7 (BMP-7) signaling. In this study, the factors that promote transcription of the longer alternate form were identified. NF-κB was shown to be important for expression of the long HuR mRNA, as was a newly identified region with potential for binding the Sp/KLF families of transcription factors. -

Podocyte Specific Knockdown of Klf15 in Podocin-Cre Klf15flox/Flox Mice Was Confirmed
SUPPLEMENTARY FIGURE LEGENDS Supplementary Figure 1: Podocyte specific knockdown of Klf15 in Podocin-Cre Klf15flox/flox mice was confirmed. (A) Primary glomerular epithelial cells (PGECs) were isolated from 12-week old Podocin-Cre Klf15flox/flox and Podocin-Cre Klf15+/+ mice and cultured at 37°C for 1 week. Real-time PCR was performed for Nephrin, Podocin, Synaptopodin, and Wt1 mRNA expression (n=6, ***p<0.001, Mann-Whitney test). (B) Real- time PCR was performed for Klf15 mRNA expression (n=6, *p<0.05, Mann-Whitney test). (C) Protein was also extracted and western blot analysis for Klf15 was performed. The representative blot of three independent experiments is shown in the top panel. The bottom panel shows the quantification of Klf15 by densitometry (n=3, *p<0.05, Mann-Whitney test). (D) Immunofluorescence staining for Klf15 and Wt1 was performed in 12-week old Podocin-Cre Klf15flox/flox and Podocin-Cre Klf15+/+ mice. Representative images from four mice in each group are shown in the left panel (X 20). Arrows show colocalization of Klf15 and Wt1. Arrowheads show a lack of colocalization. Asterisk demonstrates nonspecific Wt1 staining. “R” represents autofluorescence from RBCs. In the right panel, a total of 30 glomeruli were selected in each mouse and quantification of Klf15 staining in the podocytes was determined by the ratio of Klf15+ and Wt1+ cells to Wt1+ cells (n=6 mice, **p<0.01, unpaired t test). Supplementary Figure 2: LPS treated Podocin-Cre Klf15flox/flox mice exhibit a lack of recovery in proteinaceous casts and tubular dilatation after DEX administration. -

Mediator of DNA Damage Checkpoint 1 (MDC1) Is a Novel Estrogen Receptor Co-Regulator in Invasive 6 Lobular Carcinoma of the Breast 7 8 Evelyn K
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.16.423142; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Running Title: MDC1 co-regulates ER in ILC 2 3 Research article 4 5 Mediator of DNA damage checkpoint 1 (MDC1) is a novel estrogen receptor co-regulator in invasive 6 lobular carcinoma of the breast 7 8 Evelyn K. Bordeaux1+, Joseph L. Sottnik1+, Sanjana Mehrotra1, Sarah E. Ferrara2, Andrew E. Goodspeed2,3, James 9 C. Costello2,3, Matthew J. Sikora1 10 11 +EKB and JLS contributed equally to this project. 12 13 Affiliations 14 1Dept. of Pathology, University of Colorado Anschutz Medical Campus 15 2Biostatistics and Bioinformatics Shared Resource, University of Colorado Comprehensive Cancer Center 16 3Dept. of Pharmacology, University of Colorado Anschutz Medical Campus 17 18 Corresponding author 19 Matthew J. Sikora, PhD.; Mail Stop 8104, Research Complex 1 South, Room 5117, 12801 E. 17th Ave.; Aurora, 20 CO 80045. Tel: (303)724-4301; Fax: (303)724-3712; email: [email protected]. Twitter: 21 @mjsikora 22 23 Authors' contributions 24 MJS conceived of the project. MJS, EKB, and JLS designed and performed experiments. JLS developed models 25 for the project. EKB, JLS, SM, and AEG contributed to data analysis and interpretation. SEF, AEG, and JCC 26 developed and performed informatics analyses. MJS wrote the draft manuscript; all authors read and revised the 27 manuscript and have read and approved of this version of the manuscript. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

View PDF Version
RSC Advances View Article Online PAPER View Journal | View Issue The trans/cis photoisomerization in hydrogen bonded complexes with stability controlled by Cite this: RSC Adv.,2018,8, 23698 substituent effects: 3-(6-aminopyridin-3-yl) acrylate case study† a a b a Adam Kwiatkowski, Beata Je˛drzejewska, Marek Jozefowicz,´ Izabela Grela c and Borys Osmia´ łowski * The association of aminopyridine-based acrylic acid and its salt was studied by NMR titration experiments. The AA (acceptor, acceptor) hydrogen-bonding pattern present in the salt forms a complex readily with a DD (donor, donor) hydrogen-bonding pattern of the substituted ureas even in polar and competitive environment. The double carbon–carbon bond in the acrylic acid derivative is subjected to photoisomerization. This is dependent on the association with substituted urea derivatives. The substituent in ureas influences the trans/cis isomerization kinetics and position of the photostationary Creative Commons Attribution-NonCommercial 3.0 Unported Licence. Received 9th April 2018 state. Two mechanisms that influence the photoisomerization were proposed. To the best of our Accepted 19th June 2018 knowledge, the trans/cis photoisomerization influenced by the substituent in such a hydrogen-bonding DOI: 10.1039/c8ra03042a pattern has not observed previously. It was shown that interaction with urea derivatives causes lowering rsc.li/rsc-advances of the trans-to-cis photoreaction rates. Introduction also in crystals15). Taking the proton transfer process into account some general remarks should be made. First, it is worth The urea moiety is one of the most popular supramolecular mentioning that carboxylic acids are self-complementary This article is licensed under a synthons.1–3 It forms double hydrogen-bonding but this feature (Fig. -

AMPK, Mitochondrial Function, and Cardiovascular Disease
International Journal of Molecular Sciences Review AMPK, Mitochondrial Function, and Cardiovascular Disease Shengnan Wu * and Ming-Hui Zou Center for Molecular and Translational Medicine, Georgia State University, Atlanta, GA 30303, USA; [email protected] * Correspondence: [email protected] Received: 11 June 2020; Accepted: 6 July 2020; Published: 15 July 2020 Abstract: Adenosine monophosphate-activated protein kinase (AMPK) is in charge of numerous catabolic and anabolic signaling pathways to sustain appropriate intracellular adenosine triphosphate levels in response to energetic and/or cellular stress. In addition to its conventional roles as an intracellular energy switch or fuel gauge, emerging research has shown that AMPK is also a redox sensor and modulator, playing pivotal roles in maintaining cardiovascular processes and inhibiting disease progression. Pharmacological reagents, including statins, metformin, berberine, polyphenol, and resveratrol, all of which are widely used therapeutics for cardiovascular disorders, appear to deliver their protective/therapeutic effects partially via AMPK signaling modulation. The functions of AMPK during health and disease are far from clear. Accumulating studies have demonstrated crosstalk between AMPK and mitochondria, such as AMPK regulation of mitochondrial homeostasis and mitochondrial dysfunction causing abnormal AMPK activity. In this review, we begin with the description of AMPK structure and regulation, and then focus on the recent advances toward understanding how mitochondrial dysfunction controls AMPK and how AMPK, as a central mediator of the cellular response to energetic stress, maintains mitochondrial homeostasis. Finally, we systemically review how dysfunctional AMPK contributes to the initiation and progression of cardiovascular diseases via the impact on mitochondrial function. Keywords: mitochondrial function; AMPK; cardiovascular disease 1. Introduction Cells constantly coordinate their metabolism to satisfy their energy needs and respond to the use of nutrients. -

Ten Commandments for a Good Scientist
Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Wageningen 2010 Thesis committee Thesis supervisors Prof. dr. ir. Ivonne M.C.M. Rietjens Professor of Toxicology Wageningen University Prof. dr. Albertinka J. Murk Personal chair at the sub-department of Toxicology Wageningen University Thesis co-supervisor Dr. ir. Jacques J.M. Vervoort Associate professor at the Laboratory of Biochemistry Wageningen University Other members Prof. dr. Michael R. Muller, Wageningen University Prof. dr. ir. Huub F.J. Savelkoul, Wageningen University Prof. dr. Everardus J. van Zoelen, Radboud University Nijmegen Dr. ir. Toine F.H. Bovee, RIKILT, Wageningen This research was conducted under the auspices of the Graduate School VLAG Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Thesis submitted in fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Tuesday 14 September 2010 at 4 p.m. in the Aula Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds. Ana María Sotoca Covaleda Thesis Wageningen University, Wageningen, The Netherlands, 2010, With references, and with summary in Dutch. ISBN: 978-90-8585-707-5 “Caminante no hay camino, se hace camino al andar. Al andar se hace camino, y al volver la vista atrás se ve la senda que nunca se ha de volver a pisar” - Antonio Machado – A mi madre. -

NIH Public Access Author Manuscript Gen Comp Endocrinol
NIH Public Access Author Manuscript Gen Comp Endocrinol. Author manuscript; available in PMC 2015 February 24. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Gen Comp Endocrinol. 2014 July 1; 203: 49–59. doi:10.1016/j.ygcen.2014.03.003. Krüppel-like factors are effectors of nuclear receptor signaling Joseph R. Knoedler1 and Robert J. Denver1,2 1Neuroscience Graduate Program, University of Michigan, Ann Arbor, Michigan 48109-1048 2Department of Molecular, Cellular and Developmental Biology, University of Michigan, Ann Arbor, Michigan 48109-1048 Abstract Binding of steroid and thyroid hormones to their cognate nuclear receptors (NRs) impacts virtually every aspect of postembryonic development, physiology and behavior, and inappropriate signaling by NRs may contribute to disease. While NRs regulate genes by direct binding to hormone response elements in the genome, their actions may depend on the activity of other transcription factors (TFs) that may or may not bind DNA. The Krüppel-like family of transcription factors (KLF) is an evolutionarily conserved class of DNA-binding proteins that influence many aspects of development and physiology. Several members of this family have been shown to play diverse roles in NR signaling. For example, KLFs 1) act as accessory transcription factors for NR actions, 2) regulate expression of NR genes, and 3) as gene products of primary NR response genes function as key players in NR-dependent transcriptional networks. In mouse models, deletion of different KLFs leads to aberrant transcriptional and physiological responses to hormones, underscoring the importance of these proteins in the regulation of hormonal signaling. -

Estradiol Stimulates Mitochondrial Biogenesis and Adiponectin Expression in Skeletal Muscle
G CAPLLONCH-AMER and others Mitochondrial biogenesis 221:3 391–403 Research stimulation in WGM Estradiol stimulates mitochondrial biogenesis and adiponectin expression in skeletal muscle Gabriela Capllonch-Amer1, Miquel Sbert-Roig1, Bel M Galme´s-Pascual1, Ana M Proenza1,2, Isabel Llado´1,2, Magdalena Gianotti1,2 and Francisco J Garcı´a-Palmer1,2 1Grup Metabolisme Energe` tic i Nutricio´ , Departament de Biologia Fonamental i Cie` ncies de la Salut, Institut Correspondence Universitari d’Investigacio´ en Cie` ncies de la Salut (IUNICS), Universitat de les Illes Balears, Ctra. Valldemossa, should be addressed km 7,5. E-07122 Palma de Mallorca, Illes Balears, Spain to M Gianotti 2Centro de Investigacio´ n Biome´ dica en Red Fisiopatologı´a de la Obesidad y la Nutricio´ n (CIBERobn, CB06/03/0043), Email Instituto de Salud Carlos III, Madrid, Spain [email protected] Abstract Sexual dimorphism has been found in mitochondrial features of skeletal muscle, with female Key Words rats showing greater mitochondrial mass and function compared with males. Adiponectin is an " 17b-estradiol insulin-sensitizing adipokine whose expression has been related to mitochondrial function and " testosterone that is also expressed in skeletal muscle, where it exerts local metabolic effects. The aim of this " ovariectomy research was to elucidate the role of sex hormones in modulation of mitochondrial function, as " mitochondrial biogenesis well as its relationship with adiponectin production in rat skeletal muscle. An in vivo study with " mitochondrial dynamics ovariectomized Wistar rats receiving or not receiving 17b-estradiol (E2)(10mg/kg per 48 h for " skeletal muscle Journal of Endocrinology 4 weeks) was carried out, in parallel with an assay of cultured myotubes (L6E9) treated with " adiponectin E2 (10 nM), progesterone (Pg; 1 mM), or testosterone (1 mM). -

Single Cell Regulatory Landscape of the Mouse Kidney Highlights Cellular Differentiation Programs and Disease Targets
ARTICLE https://doi.org/10.1038/s41467-021-22266-1 OPEN Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets Zhen Miao 1,2,3,8, Michael S. Balzer 1,2,8, Ziyuan Ma 1,2,8, Hongbo Liu1,2, Junnan Wu 1,2, Rojesh Shrestha 1,2, Tamas Aranyi1,2, Amy Kwan4, Ayano Kondo 4, Marco Pontoglio 5, Junhyong Kim6, ✉ Mingyao Li 7, Klaus H. Kaestner2,4 & Katalin Susztak 1,2,4 1234567890():,; Determining the epigenetic program that generates unique cell types in the kidney is critical for understanding cell-type heterogeneity during tissue homeostasis and injury response. Here, we profile open chromatin and gene expression in developing and adult mouse kidneys at single cell resolution. We show critical reliance of gene expression on distal regulatory elements (enhancers). We reveal key cell type-specific transcription factors and major gene- regulatory circuits for kidney cells. Dynamic chromatin and expression changes during nephron progenitor differentiation demonstrates that podocyte commitment occurs early and is associated with sustained Foxl1 expression. Renal tubule cells follow a more complex differentiation, where Hfn4a is associated with proximal and Tfap2b with distal fate. Mapping single nucleotide variants associated with human kidney disease implicates critical cell types, developmental stages, genes, and regulatory mechanisms. The single cell multi-omics atlas reveals key chromatin remodeling events and gene expression dynamics associated with kidney development. 1 Renal, Electrolyte, and Hypertension Division, Department of Medicine, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA. 2 Institute for Diabetes, Obesity, and Metabolism, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA, USA. -

2006 Kyoto, Japan
October 28 - November 2, 2006 ~ Kyoto, Japan ~ Final Program Table of Contents Welcome . .2 Acknowledgements . .3 Organization . .6 MDS .Committees .& Task. .Forces . .9 International .Congress .Registration .and Venue. .12 International .Congress .Information . 13-15 . Continuing .Medical .Education . .13 . Evaluations . .14 . Press .Room . .15 Program-at-a-Glance . .17 Scientific Session Definitions . .19 Scientific .Sessions . .21 Faculty . .51 Committee .& Task. .Force .Meetings . .56 Exhibitor .Information . .57 Exhibitor .Directory . .58 Floor .Plans . 62-64 Map .of .Kyoto . .66 Lunch Map . .67 Subway Map . .68 Social Events . .69 Poster .Session .1 . .72 Poster .Session .2 . .88 Poster .Session .3 . .102 Poster .Session .4 . .117 CME .Request .Form . .133 The Movement. .Disorder .Society’s 0th International Congress of Parkinson’s Disease and Movement Disorders Welcome Letter Dear Colleagues, On behalf of The Movement Disorder Society (MDS), we are pleased to welcome you to Kyoto, Japan for the 10th International Congress of Parkinson’s Disease and Movement Disorders . The 10th International Congress has been designed to provide an innovative and comprehensive overview of the latest perspectives and research developments in the field of Movement Disorders . We encourage you to take every opportunity to participate in the Scientific Program which has drawn world renowned speakers and foremost experts in their respective fields . In the next days, the latest research regarding Movement Disorders will be presented and discussed in an open format, offering unique educational opportunities for all delegates . The International Congress convenes with a series of Opening Seminars and then continues with an array of Plenary, Parallel, Poster and Video Sessions, as well as Lunch Seminars, Controversies and Skills Workshops .