Thesis, and Not to Quote Or Make a Copy of the Whole Or Any Substantial Part of It Without the Written Consent of the Author and of the University of Bath
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Parish News March 2020
Chew Stoke with Nempnett Thrubwell Parish News [email protected] Free for EVERYONE in the area March 2020 https://lakesidegroup.org.uk/churches 1 Pilates Classes run by Emma Charlton at AVAILABLE FOR HIRE Suitable for private parties, Butcombe Village Hall group activities etc. Regular Hire / Single Event Hire Pilates is an exercise & it is Modern kitchen and toilet facilities, suitable for most people. bar area and stage Pilates can help you to to see information about the hall, to view our calendar for availability and to book improve your flexibility, help you gain phone 07583 289965 or online strength & help you relax. [email protected] The classes are small & friendly chewstokechurchhall.org.uk Please contact me if you would like to come along to a class. [email protected] NEMPNETT THRUBWELL VILLAGE HALL is situated in a beautiful rural location with amazing views! It is an ideal venue for all types of functions and occasions, having extensive parking, level access, full disabled facilities, fully fitted kitchen, video/audio system and children’s play area.. To book or for more details, please visit our Facebook page or call 01761 462391 2 3 4 MICHAEL W. ROWE FUNERAL DIRECTOR A family business offering personal service day or night Rest Room facilities 2 The Vinery, Harford Square Chew Magna, BS40 8RD [email protected] Telephone: 01275 332565 5 6 7 ******* USEFUL CONTACTS ****** Chew Stoke Parish Council Chairperson: Dick Raffety 01275 331102 Clerk: Catherine Parkman 07779 592328 Vice– Chairperson: -

THE DEEPINGS ADVERTISER Ideal
16th Oct Deepings & Villages THE DEEPINGS ADVERTISER Advertiser Tel (01778) 348319 : [email protected] : www.the-advertiser.co.uk : Facebook - DeepingsAdvertiser 2020 Result Carpentry & Joinery Cloud 9 Decor A well-established business, with over 25 þ All External Renders ELECTRICIAN For all your years experience, offering high quality Local, Qualified workmanship. þ All Plastering Requirements 18th edition Painting & Decorating needs Full carpentry services available. T: 01778 560292 M: 07860 817 415 For a prompt 01778 343676 Contact Joe on High end results without the high end costs. FREE quote or Contact Gary 07787 124 107 www.albysplastering.co.uk call Tony on: 07813 872762 07517 227 038 Website: resultcarpentry.com Computer Problems? • Timber fencing supplied and fitted • NO FIX NO FEE with concrete or wooden posts No job • NO CALL OUT CHARGE • FIXED FEES • Gates made to measure too small • NO QUIBBLE FOLLOW-UPS For a FREE quote call 01733 767 276 or Mob 07765 268 441 • PICK UP / DROP OFF Email: [email protected] • AVAILABLE 7 DAYS A WEEK • HOME & BUSINESS SUPPORT Michael Mitchell Plumbing & Handyman Established for over 30 years 01778 485551 Based in Deeping St James www.idealitservices.co.uk ideal I T S E R V I C E S –Bathrooms –Tiling [email protected] HIGH QUALITY, LOW COST IT SUPPORT –Household Plumbing (not gas certified) –Repairs –Patio Power Cleaning –Shower Steam Cleaning –General Maintenance No job too small 07815 953 833 / [email protected] Message from Phil Dilks, District councillor for Deeping St James Please help 'Save Back Lane' and support local residents. -

October 2020 at 7:00Pm
The Institute, 38 Church Street, Deeping St James, Peterborough PE6 8HD E-mail: [email protected] Tel: 01778 343266 //parishes.lincolnshire.gov.uk/deepingstjames Parish Clerk: Julie Fortnum Minutes of the virtual Planning & Transport Committee held on Thursday 22 October 2020 at 7:00pm. Present: Councillors Gilbert, Shinkins-Hoppe, Lilley, Hosking, Stevens, Thomas and District Councillor Dilks. Julie Fortnum, Parish Clerk and Louise Brown, Assistant Clerk. 44.21 To receive apologies of absence. Apologies were received from Councillor Hardy. 45.21 To receive declarations of interest, if any. None received. 46.21 To agree the Minutes of the previous meeting held on 17th September 2020 The minutes were agreed as a true representation and will be signed. 47.21 To receive an update regarding Planning Application S20/0632 Appreciation was offered to both District Councillor Dilks and Councillor Shinkins-Hoppe for their dealings with this case. It appeared that there were various inconsistencies throughout the hearing of this application and Councillor Dilks welcomed a response from the Council outlining their concerns. Councillor Shinkins-Hoppe offered to put together a letter for consideration at Full Council and would recommended this be sent on behalf of the Parish Council. 48.21 To receive reports from the clerk. Nothing to report. 49.21 To offer a response to the Government White Paper – Planning for the Future. The Councillors were given two appendices supplied by Councillor Shinkins-Hoppe. The first report gave a shortened explanation of the proposed changes to Planning Law. The second document represented the Parish Council’s response to the questions raised within the White Paper. -

Sat 14Th and Sun 15Th October 2017 10Am To
CHEW VALLEY BLAGDON BLAGDON AND RICKFORD RISE, BURRINGTON VENUE ADDRESSES www.chewvalleyartstrail.co.uk To Bishopsworth & Bristol Sarah Jarrett-Kerr Venue 24 Venue 11 - The Pelican Inn, 10 South Margaret Anstee Venue 23 Dundry Paintings, mixed media and prints Book-binding Parade, Chew Magna. BS40 8SL North Somerset T: 01761 462529 T: 01761 462543 Venue 12 - Bridge House, Streamside, E: [email protected] E: [email protected] Chew Magna. BS40 8RQ Felton Winford Heights 2 The art of seeing means everything. The wonderful heft and feel of leather To A37 119 7 Landscape and nature, my inspiration. bound books and journals. Venue 13 - Longchalks, The Chalks, Bristol International Pensford B3130 3 & Keynsham Chew Magna. BS40 8SN Airport 149Winford Upton Lane Suzanne Bowerman Venue 23 Jeff Martin Venue 25 Sat 14th and Sun 15th Venue 14 - Chew Magna Baptist Chapel, Norton Hawkfield Belluton Paintings Watercolour painting A38 T: 01761 462809 Tunbridge Road, Chew Magna. BS40 8SP B3130 October 2017 T: 0739 9457211 Winford Road B3130 E: [email protected] E: [email protected] Venue 15 - Stanton Drew Parish Hall, Sandy 192 13 1S95tanton Drew Colourful, atmospheric paintings in a To Weston-Super-Mare 17 An eclectic mix of subjects - landscapes, 5 11 16 10am to 6pm variety of subjects and mediums. Lane, Stanton Drew. BS39 4EL or Motorway South West 194 seascapes, butterflies, birds and still life. Regil Chew Magna CV School Venue 16 - The Druid's Arms, 10 Bromley Stanton Wick Chris Burton Venue 23 Upper Strode Chew Stoke 8 VENUE ADDRESSES Road, Stanton Drew. BS39 4EJ 199 Paintings 6 Denny Lane To Bath T: 07721 336107 Venue 1 - Ivy Cottage, Venue 17 - Alma House, Stanton Drew, (near A368 E: [email protected] 50A Stanshalls Lane, Felton. -

Robert Wait (Alias Wayte) Rachel (Daniels Or Ford)
Robert Wait (alias Wayte) 1695 - 1755 Farmer of Rangeworthy and Iron Acton in Gloucestershire, and of Norton Malreward in Somerset AND HIS FIRST WIFE Rachel (Daniels or Ford) ? - 1747 by Geoffrey Audcent © 2014, Mendip Road, Yatton, North Somerset William Wait (1669-1754) Farmer of Castle Combe Robert Wait (1695-1755) Farmer of Rangeworthy and Norton Malreward Abstract – Robert Wait was born and baptised at Castle Combe in Wiltshire in 1695 (os). He was a successful farmer at Rangeworthy and Iron Acton (where he was the tenant of Acton House Farm) in Gloucestershire, before moving to northern Somerset in 1744, where he took over a large farm at Norton Malreward. Robert had nine children by his first wife. She was an early Methodist and travelled to Kingswood every Sunday to hear the Methodist ministers who preached there. Following her death in 1747 Robert remarried and had one further child. In 1749 Robert made a will, describing himself as a farmer of the parish of Norton Maleward, in good health and in perfect mind and memory “thanks be given to God therefore”. Robert died in 1755 and was buried at Norton Malreward. His will was proved in London at the Prerogative Court of Canterbury in July 1755 with administration of all goods, chattels and credits granted to his eldest son. Robert Wait (alias Wayte) 1695 - 1755 Farmer of Rangeworthy and Iron Acton in Gloucestershire, and of Norton Malreward in Somerset AND HIS FIRST WIFE Rachel (Daniels or Ford) ? - 1747 Six-Times-Great-Grandparents to Mrs Sandra Audcent (nee Russell) Robert Waite was born on 19 January 1695 at Castle Combe in Wiltshire (the “prettiest village in England”).1 He was baptised there in the parish church of St. -
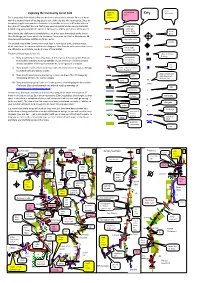
Communityforestpath.Pdf
1 – Clifton Suspension Bridge to Colliter’s Brook and Bedminster 2 – Up to Dundry Down – Buses 8, (9) 486 and 487 at start, 75 (or 25) at finish – Buses 75 (or 25) at start 75 (or 30) at finish Access route via number 8 bus from Temple Meads or the Centre to Christchurch, Clifton. (Number 9 bus makes the same trip in the opposite direction, which involves a diversion I have never really understood why so many people have problems with this through Cotham.) 486 and 487 buses also stop at Christchurch and link to Kingswood and section, but the fact that do should warn you to study this plan and the OS map north central Bristol. return to Centre using 75 bus. carefully. It could be that the tower of Dundry Parish Church lures people off course, or perhaps they haven’t got their eye in yet! From number 8 bus stop, cross green to start point past toilets near Clifton Suspension Bridge where Community Forest Path (CFP) descends from Observatory past the sliding The reliable 75 bus links either end of this section. The 75 also serves the rock known as the Tailor’s Friend. Challenge route in Patchway, which means that you can access the whole 15 mile This is the most complicated map of all due to the fact that there are no officially western section of the route using this bus. The closest point for me to access recognised ‘Public Rights of Way’ through the Ashton Court Estate, which means that the the 75 is near the Wellington hotel on Horfield Common, which suggests an lodge gates are shut at night, a problem for a 24-hour challenge. -

7286 the London Gazette, 10 November, 1933
7286 THE LONDON GAZETTE, 10 NOVEMBER, 1933 DISEASES OF ANIMALS ACTS, In the county of Cambridge. 1894 TO 1927. The parishes of Great Chishill, Little Chis- MINISTEY OF AGRICULTURE AND FISHERIES. hall and Heydon. Notice is hereby given, in pursuance of Section 49 (3) of the Diseases of Animals Act, In the county of Essex. 1894, that the Minister of Agriculture and The parish of Chrishall (except its detached Fisheries has made the following Orders. part). Order No. 5165. (ii) Further contraction of the Isle of Ely (Dated 6th November, 1933). Foot-and-Mouth Disease Infected Area. FOOT-AND-MOUTH DISEASE (INFECTED Substitutes on the 13th November, 1933, the AREAS) ORDER OF 1933 (No. 81). following Area for the Infected Area described in the Second Schedule to the Foot-and-Mouth SUBJECT. Disease (Infected Areas) Order of 1933 (No. Contraction of the Somerset Foot-and-Mouth 78):— Disease Infected Area. An Area comprising: — Substitutes on the llth November, 1933, the following Area for the Infected Area described In the counties of Cambridge and the Isle of in the Schedule to the Foot-and-Mouth Disease Ely. (Infected Areas) Order of 1933 (No. 77):— So much of the Parishes of Waterbeach, An Area comprising:— Swaffham Bulbeck, Swaffham Prior, Burwell, Wicken and Stretham as lies within the follow- In the county of Somerset. ing boundary, namely:— The petty sessional divisions of Long Ashton Commencing at Stretham Ferry Bridge on (except the parish of Kingston Seymour) and the main Cambridge—Ely road; thence in a Keynsham. north-easterly direction -

Area 1: Thrubwell Farm Plateau
Area 1: Thrubwell Farm Plateau Summary of Landscape Character • Clipped hedges which are often ‘gappy’ and supplemented by sheep netting • Late 18th and early 19th century rectilinear field layout at north of area • Occasional groups of trees • Geologically complex • Well drained soils • Flat or very gently undulating plateau • A disused quarry • Parkland at Butcombe Court straddling the western boundary • Minor roads set out on a grid pattern • Settlement within the area consists of isolated farms and houses For detailed Character Area map see Appendix 3 23 Context Bristol airport on the plateau outside the area to the west. Introduction Land-uses 7.1.1 The character area consists of a little over 1sq 7.1.6 The land is mainly under pasture and is also km of high plateau to the far west of the area. The plateau used for silage making. There is some arable land towards extends beyond the Bath and North East Somerset boundary the north of the area. Part of Butcombe Court parkland into North Somerset and includes Felton Hill to the north falls within the area to the west of Thrubwell Lane. and Bristol airport to the west. The southern boundary is marked by the top of the scarp adjoining the undulating Fields, Boundaries and Trees and generally lower lying Chew Valley to the south. 7.1.7 Fields are enclosed by hedges that are generally Geology, Soils and Drainage trimmed and often contain few trees. Tall untrimmed hedges are less common. Hedges are typically ‘gappy’ and of low 7.1.2 Geologically the area is complex though on the species diversity and are often supplemented with sheep- ground this is not immediately apparent. -

Gjland at NORTON MALREWARD
Figure 1POUND COTTAGE, ALHAMPTON gjLAND ATPITCOT COTTAGE NORTONSTRATTON MALREWARD ON THE FOSSE PITCOT COTTAGE, STRATTON ON THE FOSSE, SOMERSET Wells 12 miles • Bath 12 miles • Bristol 16 miles • Frome 10 miles Guide Price £715,000 A detached residential dwelling having been extended over the years to now offer balanced accommodation set in its own landscaped gardens with stunning views over the Somerset countryside and approx. 12 acres with stabling. Kitchen Three Double Bedrooms all with Ample Parking Ensuite Facilities Dining Room Previous Planning Permission Study/Fourth Bedroom Sitting Room for conversion of the loft with Detached Triple Garage two dormer windows. Cloakroom Landscaped Gardens of approx. Stabling with approx. 12 acres Utility Room 1.5 acres DESCRIPTION Pitcot is a spacious family home with well-appointed accommodation and set in approximately one acre of landscaped gardens. The kitchen is south facing with french doors leading out to the balcony dining area. The sitting room is triple aspect resulting in a light and airy living space. An open fireplace with a decorative cast surround and wooden mantle provides a central point to the spacious room. Another access to the balcony is through the french doors in the dining room this providing excellent views whilst dining. The study/fourth bedroom is also located on this floor along with a utility. There are three further double bedrooms all with ensuite facilities. The property has previous planning permission for a fifth bedroom suite. The property is approached off the lane via a five bar ranch style gate which leads to the sweeping driveway. To the right hand side of the driveway is approx. -

Gjland at NORTON MALREWARD
Figure 1POUND COTTAGE, ALHAMPTON gjLAND AT 8 INNICKS CLOSE NORTON MALREWARDUBLEY 8 INNCIKS CLOSE, UBLEY, NEAR BRISTOL Bristol 16 miles • Bath 18 • Wells 12 miles Guide Price £349,950 A three bedroom semi-detached house that has been the cherished home of the current vendors for the past 21 years. The property is located in the popular village of Ubley surrounded by beautiful rural countryside. Sitting Room Three Bedrooms Picturesque Countryside Views Kitchen/ Dining Room Popular Location Quiet and Peaceful Utility Area Corner Plot Potential to Extend Beautiful Large Garden Easy access to Bristol, Bath and Wells Cloakroom DESCRIPTION Tucked away in the quiet village of Ubley, 8 Innicks Close is a three bedroom semi-detached house that has great potential to extend and make a wonderful home for any potential buyer. The property boasts countryside views over the Mendips and fronts onto the village green. On arriving to the property, you enter into the hallway. To the left you have the sitting room, a bright room which leads you to the conservatory which overlooks the back garden. The dual aspect kitchen/ dining room benefits from plenty of storage space and has access to the pantry, utility area and downstairs cloakroom. Upstairs there are two double bedrooms, one single bedroom and a family bathroom, fitted with a shower, toilet and wash hand basin. With the property being a corner plot, 8 Innicks Close has a very large garden compared to the other properties in the close. The garden sweeps along the side of the property and has been well cared for by the current vendors. -

Full Property Address Account Start Date
Property Reference Number Name (Redacted as Personal Data if Blank) Full Property Address Account Start Date 10010080460 46, Alexandra Road, Grantham, Lincolnshire, NG31 7AP 01/04/2005 10010080463 Lincolnshire County Council Lincs County Council, Alexandra Road, Grantham, Lincolnshire, NG31 7AP 01/04/2005 10010160350 Avc 35 Ltd The Avenue Veterinary Centre, 35, Avenue Road, Grantham, Lincolnshire, NG31 6TA 01/04/2005 10010615050 Neat Ideas Ltd Unit 5, Belton Lane Industrial Estate, Belton Lane, Grantham, Lincolnshire, NG31 9HN 01/04/2005 10010695200 8, Bridge Street, Grantham, Lincolnshire, NG31 9AE 01/04/2005 10010710010 2nd Grantham(St Wulframs) Scouts Group 2nd Grantham Scout Group, Broad Street, Grantham, Lincolnshire, NG31 8AP 01/04/2005 10010720340 The Board Of Governors The Kings School The Kings School, Brook Street, Grantham, Lincolnshire, NG31 6PS 01/04/2005 10011150140 14, Castlegate, Grantham, Lincolnshire, NG31 6SE 01/04/2005 10011150160 16, Castlegate, Grantham, Lincolnshire, NG31 6SE 01/04/2005 10011150500 Grantham Conservative Club 50, Castlegate, Grantham, Lincolnshire, NG31 6SN 01/04/2005 10011150660 The Castlegate, 69, Castlegate, Grantham, Lincolnshire, NG31 6SJ 01/04/2005 10011290453 The Maltings Dental Practice The Maltings, Commercial Road, Grantham, Lincolnshire, NG31 6DE 01/04/2005 10011300272 South Kesteven District Council South Kesteven District Council, Conduit Lane, Grantham, Lincolnshire, NG31 6LQ 01/04/2005 10011810010 Dudley House School 1, Dudley Road, Grantham, Lincolnshire, NG31 9AA 01/04/2005 10011820020 -

Corre Strategy
North Somerset Council Local Development Framework Core Strategy Topic paper Settlement Function and Hierarchy September 2007 Settlement Function and Hierarchy This is part of a series of topic papers summarising the evidence base for the North Somerset Core Strategy document. Other topic papers available in this series: Demography, health, social inclusion and deprivation Housing Economy Retail Leisure, Tourism and Culture Resources (including minerals, waste, recycling, energy consumption) Natural environment (including climate change, biodiversity, green infrastructure, countryside, natural environment and flooding) Transport and communications Sustainable construction / design quality including heritage Summing up / spatial portrait For further information on this topic paper please contact: Planning Policy Team Development and Environment North Somerset Council Somerset House Oxford Street Weston-super-Mare BS23 1TG Tel: 01275 888545 Fax: 01275 888569 [email protected] 2 1.0 Introduction 1.1 The functional relationship particularly between where people choose to live and work is no longer as closely related as in the past. The relationship can be quite complex. It is determined by numerous factors which need to be taken into account in planning for growth. 1.2 The purpose of this Topic Paper is to set out the evidence in terms of the settlements in North Somerset in order to identify their current function and functional relationships with other settlements. The evidence will form the basis on which a hierarchy of settlements will be established. In particular the identification of Development Policy B and C settlements set out in the draft RSS. The Paper will also consider how policies have influenced the form and function of settlements and inform any discussion of whether the current list of inset and washed over Green Belt settlements is still appropriate and whether settlement boundaries are still the appropriate planning tool for many green belt and rural settlements.