Fusion Genes and Rnas in Cancer Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cellular and Synaptic Network Defects in Autism
Cellular and synaptic network defects in autism The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Peca, Joao, and Guoping Feng. “Cellular and Synaptic Network Defects in Autism.” Current Opinion in Neurobiology 22, no. 5 (October 2012): 866–872. As Published http://dx.doi.org/10.1016/j.conb.2012.02.015 Publisher Elsevier Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/102179 Terms of Use Creative Commons Attribution-Noncommercial-NoDerivatives Detailed Terms http://creativecommons.org/licenses/by-nc-nd/4.0/ NIH Public Access Author Manuscript Curr Opin Neurobiol. Author manuscript; available in PMC 2013 October 01. Published in final edited form as: Curr Opin Neurobiol. 2012 October ; 22(5): 866–872. doi:10.1016/j.conb.2012.02.015. Cellular and synaptic network defects in autism João Peça1 and Guoping Feng1,2 $watermark-text1McGovern $watermark-text Institute $watermark-text for Brain Research, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge, MA 02139, USA 2Stanley Center for Psychiatric Research, Broad Institute, Cambridge, MA 02142, USA Abstract Many candidate genes are now thought to confer susceptibility to autism spectrum disorder (ASD). Here we review four interrelated complexes, each composed of multiple families of genes that functionally coalesce on common cellular pathways. We illustrate a common thread in the organization of glutamatergic synapses and suggest a link between genes involved in Tuberous Sclerosis Complex, Fragile X syndrome, Angelman syndrome and several synaptic ASD candidate genes. When viewed in this context, progress in deciphering the molecular architecture of cellular protein-protein interactions together with the unraveling of synaptic dysfunction in neural networks may prove pivotal to advancing our understanding of ASDs. -

Identification and Characterization of a Novel 43-Bp Deletion Mutation of The
Liu et al. BMC Medical Genetics (2018) 19:61 https://doi.org/10.1186/s12881-018-0567-z CASE REPORT Open Access Identification and characterization of a novel 43-bp deletion mutation of the ATP7B gene in a Chinese patient with Wilson’s disease: a case report Gang Liu†, Dingyuan Ma†, Jian Cheng, Jingjing Zhang, Chunyu Luo, Yun Sun, Ping Hu, Yuguo Wang, Tao Jiang* and Zhengfeng Xu* Abstract Background: Wilson’s disease (WD) is an autosomal recessive disorder characterized by copper accumulation. ATP7B gene mutations lead to ATP7B protein dysfunction, which in turn causes Wilson’s disease. Case presentation: We describe a male case of Wilson’s disease diagnosed at 10 years after routine biochemical test that showed low serum ceruloplasmin levels and Kayser–Fleischer rings in both corneas. Analysis of the ATP7B gene revealed compound heterozygous mutations in the proband, including the reported c.3517G > A mutation and a novel c.532_574del mutation. The c.532_574del mutation covered a 43-bp region in exon 2, and resulted in a frameshift mutation (p.Leu178PhefsX10). By base sequence analysis, two microhomologies (TCTCA) were observed on both deletion breakpoints in the ATP7B gene. Meanwhile, the presence of some sequence motifs associated with DNA breakage near the deletion region promoted DNA strand break. Conclusions: By comparison, a replication-based mechanism named fork stalling and template switching/ microhomology-mediated break-induced replication (FoSTeS/MMBIR) was used to explain the formation of this novel deletion mutation. Keywords: Wilson’s disease, ATP7B, Novel mutation, FoSTeS/MMBIR Background ATP7B protein dysfunction, which in turn causes accumu- Wilson’s disease (WD, OMIM #277900), an autosomal lation of copper in the liver, brain, kidneys and corneas, recessive disorder characterized by abnormal copper ac- with a wide range of clinical symptoms, including hepatic cumulation and related toxicities, is caused by mutations disorders, neuronal degeneration of the brain, and Kayser- in the ATP7B gene (OMIM *606882) [1]. -

Retrocopy Contributions to the Evolution of the Human Genome Robert Baertsch*1, Mark Diekhans1, W James Kent1, David Haussler1 and Jürgen Brosius2
BMC Genomics BioMed Central Research article Open Access Retrocopy contributions to the evolution of the human genome Robert Baertsch*1, Mark Diekhans1, W James Kent1, David Haussler1 and Jürgen Brosius2 Address: 1Center for Biomolecular Science and Engineering, University of California Santa Cruz, Santa Cruz, California 95064, USA and 2Institute of Experimental Pathology, ZMBE, University of Münster, Von-Esmarch-Str. 56, D-48149, Münster, Germany Email: Robert Baertsch* - [email protected]; Mark Diekhans - [email protected]; W James Kent - [email protected]; David Haussler - [email protected]; Jürgen Brosius - [email protected] * Corresponding author Published: 8 October 2008 Received: 17 March 2008 Accepted: 8 October 2008 BMC Genomics 2008, 9:466 doi:10.1186/1471-2164-9-466 This article is available from: http://www.biomedcentral.com/1471-2164/9/466 © 2008 Baertsch et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Evolution via point mutations is a relatively slow process and is unlikely to completely explain the differences between primates and other mammals. By contrast, 45% of the human genome is composed of retroposed elements, many of which were inserted in the primate lineage. A subset of retroposed mRNAs (retrocopies) shows strong evidence of expression in primates, often yielding functional retrogenes. Results: To identify and analyze the relatively recently evolved retrogenes, we carried out BLASTZ alignments of all human mRNAs against the human genome and scored a set of features indicative of retroposition. -

Nf1 Gene Deletion
NF1 GENE DELETION NF1 GENE DELETION This resource is for families who have a deletion of the NF1 gene causing neurofi bromatosis type 1 (NF1). This is also referred to as NF1 microdeletion. WHAT ARE CHROMOSOMES, DELETION GENES AND MUTATIONS? Chromosomes are the packages of our genetic information. Within each cell of the body are 46 chromosomes arranged in 23 pairs. One chromosome in each pair is inherited from the Source: U.S. National Library of Medicine mother and the other from the father. The pairs are numbered WHAT IS AN NF1 MICRODELETION? by size. The number 1 chromosome When the entire NF1 gene is missing, it is referred pair is the largest and the number 22 is to as NF1 gene deletion or NF1 microdeletion. the smallest. The last pair of chromosomes Approximately 5% of individuals with a diagnosis (sex chromosomes) help to determine whether of NF1 have a deletion that includes the entire NF1 an individual is a male or a female. Genes are gene. Other than the NF1 gene, there are usually small areas along the chromosomes, and are other nearby genes that are also missing. the body’s blueprints or instructions. We have approximately 20,000 genes that control how we WHAT DOES IT MEAN TO HAVE AN NF1 grow and develop and what we look like. Each MICRODELETION? gene can be thought of as a sentence made up of In addition to the NF1 gene, individuals with NF1 four letters (A, T, C and G). Mutations (also called microdeletion typically have other genes in the pathogenic variants), are changes in a gene’s region of chromosome 17 deleted. -

RNA-Dependent Amplification of Mammalian Mrna Encoding Extracellular Matrix Components
bioRxiv preprint doi: https://doi.org/10.1101/376293; this version posted December 5, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. RNA-DEPENDENT AMPLIFICATION OF MAMMALIAN mRNA ENCODING EXTRACELLULAR MATRIX COMPONENTS: IDENTIFICATION OF CHIMERIC RNA INTERMEDIATES FOR a1, b1, AND g1 CHAINS OF LAMININ. * Vladimir Volloch 1 , Sophia Rits 2,3, Bjorn Olsen 1 1: Department of Developmental Biology, Harvard School of Dental Medicine 2: Howard Hughes Medical Institute at Children’s Hospital, Boston 3: Dept. of Biological Chemistry and Molecular Pharmacology, Harvard Medical School *: Corresponding Author, [email protected]; [email protected] ABSTRACT. The aim of the present study was to test for the occurrence of key elements predicted by the previously postulated mammalian RNA-dependent mRNA amplification model in a tissue producing massive amounts of extracellular matrix proteins. At the core of RNA-dependent mRNA amplification, until now only described in one mammalian system, is the self-priming of an antisense strand and extension of its 3’ terminus into a sense-oriented RNA containing the protein-coding information of a conventional mRNA. The resulting product constitutes a new type of biomolecule. It is chimeric in that it contains covalently connected antisense and sense sequences in a hairpin configuration. Cleavage of this chimeric intermediate in the loop region of a hairpin structure releases mRNA which contains an antisense segment in its 5’UTR; depending on the position of self-priming, the chimeric end product may encode the entire protein or its C-terminal fragment. -

Variations in Microrna-25 Expression Influence the Severity of Diabetic
BASIC RESEARCH www.jasn.org Variations in MicroRNA-25 Expression Influence the Severity of Diabetic Kidney Disease † † † Yunshuang Liu,* Hongzhi Li,* Jieting Liu,* Pengfei Han, Xuefeng Li, He Bai,* Chunlei Zhang,* Xuelian Sun,* Yanjie Teng,* Yufei Zhang,* Xiaohuan Yuan,* Yanhui Chu,* and Binghai Zhao* *Heilongjiang Key Laboratory of Anti-Fibrosis Biotherapy, Medical Research Center, Heilongjiang, People’s Republic of China; and †Clinical Laboratory of Hong Qi Hospital, Mudanjiang Medical University, Heilongjiang, People’s Republic of China ABSTRACT Diabetic nephropathy is characterized by persistent albuminuria, progressive decline in GFR, and second- ary hypertension. MicroRNAs are dysregulated in diabetic nephropathy, but identification of the specific microRNAs involved remains incomplete. Here, we show that the peripheral blood from patients with diabetes and the kidneys of animals with type 1 or 2 diabetes have low levels of microRNA-25 (miR-25) compared with those of their nondiabetic counterparts. Furthermore, treatment with high glucose decreased the expression of miR-25 in cultured kidney cells. In db/db mice, systemic administration of an miR-25 agomir repressed glomerular fibrosis and reduced high BP. Notably, knockdown of miR-25 in normal mice by systemic administration of an miR-25 antagomir resulted in increased proteinuria, extracellular matrix accumulation, podocyte foot process effacement, and hypertension with renin-angiotensin system activation. However, excessive miR-25 did not cause kidney dysfunction in wild-type mice. RNA sequencing showed the alteration of miR-25 target genes in antagomir-treated mice, including the Ras-related gene CDC42. In vitro,cotrans- fection with the miR-25 antagomir repressed luciferase activity from a reporter construct containing the CDC42 39 untranslated region. -

CD29 Identifies IFN-Γ–Producing Human CD8+ T Cells With
+ CD29 identifies IFN-γ–producing human CD8 T cells with an increased cytotoxic potential Benoît P. Nicoleta,b, Aurélie Guislaina,b, Floris P. J. van Alphenc, Raquel Gomez-Eerlandd, Ton N. M. Schumacherd, Maartje van den Biggelaarc,e, and Monika C. Wolkersa,b,1 aDepartment of Hematopoiesis, Sanquin Research, 1066 CX Amsterdam, The Netherlands; bLandsteiner Laboratory, Oncode Institute, Amsterdam University Medical Center, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; cDepartment of Research Facilities, Sanquin Research, 1066 CX Amsterdam, The Netherlands; dDivision of Molecular Oncology and Immunology, Oncode Institute, The Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands; and eDepartment of Molecular and Cellular Haemostasis, Sanquin Research, 1066 CX Amsterdam, The Netherlands Edited by Anjana Rao, La Jolla Institute for Allergy and Immunology, La Jolla, CA, and approved February 12, 2020 (received for review August 12, 2019) Cytotoxic CD8+ T cells can effectively kill target cells by producing therefore developed a protocol that allowed for efficient iso- cytokines, chemokines, and granzymes. Expression of these effector lation of RNA and protein from fluorescence-activated cell molecules is however highly divergent, and tools that identify and sorting (FACS)-sorted fixed T cells after intracellular cytokine + preselect CD8 T cells with a cytotoxic expression profile are lacking. staining. With this top-down approach, we performed an un- + Human CD8 T cells can be divided into IFN-γ– and IL-2–producing biased RNA-sequencing (RNA-seq) and mass spectrometry cells. Unbiased transcriptomics and proteomics analysis on cytokine- γ– – + + (MS) analyses on IFN- and IL-2 producing primary human producing fixed CD8 T cells revealed that IL-2 cells produce helper + + + CD8 Tcells. -

Generating High Variability of B Chromosomes in Eyprepocnemis Plorans (Grasshopper)
Heredity 71 (1993) 352—362 Received 5 January 1993 Genetical Society of Great Britain Generating high variability of B chromosomes in Eyprepocnemis plorans (grasshopper) M. D. LOPEZ-LEON, J. CABRERO, M. C. PARDO, E. VISERAS, J. P. M. CAMACHO* & J. L. SANTOSI Departamento de Genética, Facu/tad de Ciencias, Un/versidad de Granada, E- 18071 Granada, Spa/n and tDepartamento de Genét/ca, Facultad de B/o/ogIa, Un/vers/dad Comp/utense de Madrid, E-28040 Madrid, Spa/n Twenty-eightprogeny analyses (PAs) performed on specimens of E. plorans collected from four natural Iberian populations have been informative about the transmission of rare B chromosome types or the de novo origin of some of them. At least ii rare B-types have been found in addition to the predominant ones: B1 in Daimuz, B2 in Jete and Salobreña, and B5 in Fuengirola. The presence in two controlled crosses of one embryo carrying a B-type which was absent in the parents suggests that these B variants (B20 and B )haveoriginated de novo. Eleven other PAs suggest that new B derivatives are recurrently arising in these populations. The most frequent B chromosome mutation was centromere misdivision that originated four different B-types (B2m1,B110,B210 and Bmjnj). Other rearrangements were pericentric inversions (B211, B212 and B213), inverse tandem fusion (B211), centric fusion (B11) and deletions (B2d1andB2d2).Thefour B derivatives produced by centromeric misdivision are significantly eliminated during sexual transmission, most probably owing to deficiencies in the control of chromosome movement by their hemicentromeres. Those derived from translocations showed Mendelian transmission but deletion B variants showed a tendency to elimination. -

Continuous Variation in Y-Chromosome Structure of Rumex Acetosa
Heredity 57 (1986) 247-254 The Genetical Society of Great Britain Received 16 December 1985 Continuous variation in Y-chromosome structure of Rumex acetosa A. S. Wilby and School of Biological Sciences, J. S. Parker Queen Mary College, Mile End Road, London El 4NS. The dioecious angiosperm Rumex acetosa has an XXIXY1Y2sex-chromosomesystem. Each V-chromosome is heterochromatic except for a minute terminal euchromatic pairing segment. The Vs are constant in size but have a variable centromere position. The centromeres can be located anywhere within the central 40 per cent of the chromosome but are excluded from the two distal 30 per cent regions. In a sample of 270 males from 18 different populations 68 distinct variants have been identified on the basis of V-morphology. All populations are highly polymorphic with a minimum of four variants in a sample of ten males. The origin and significance of this massive variability is considered in this paper. Increased mutation rate of the Ys may be implicated in maintenance of this variation. I NTRO DUCTI ON these "inert" Ys has been described (Vana, 1972) variation in their structure has been overlooked. Sex-determinationin animals is usually genic and Extensive heterochroinatic content is a charac- frequently associated with visibly-differentiated teristic of many Y- and W-chromosomes. Indeed, sex-chromosomes. Sex expression in plants, some have argued that the process of hetero- however, is usually more plastic, and is subject to chromatinisation itself was implicated in the initial environmental influences such as temperature and phase of sex-chromosome differentiation (Jones, photoperiod (Heslop-Harrison, 1957). -

Elimination Pathways of Fusion Protein and Peptide Drugs
Review Volume 11, Issue 2, 2021, 9139 - 9147 https://doi.org/10.33263/BRIAC112.91399147 Elimination Pathways of Fusion Protein and Peptide Drugs Ali Khodadoust 1 , Hossein Aghamollaei 2 , Ali Mohammad Latifi 1 , Kazem Hasanpour 3 , Mahdi Kamali 4 , Hamid Tebyanian 5 , Gholamreza Farnoosh 1,* 1 Applied Biotechnology Research Center, Baqiyatallah University of Medical Sciences, Tehran, Iran 2 Chemical Injuries Research Center, Systems biology and Poisonings Institute, Baqiyatallah University of Medical Sciences, Tehran, Iran 3 Sabzevar University of Medical Sciences, School of Medicine, Sabzevar, Iran 4 Nanobiotechnology Research Center, Baqiyatallah University of Medical Sciences, Tehran, Iran 5 Research Center for Prevention of Oral and Dental Diseases, Baqiyatallah University of Medical Sciences, Tehran, Iran * Correspondence: [email protected]; Scopus Author ID 55855454400 Received: 28.07.2020; Revised: 25.08.2020; Accepted: 27.08.2020; Published: 1.09.2020 Abstract: Fusion proteins have been known as an interesting subject for scientific researches in improving properties or making a new function by synergistically incorporating two protein domains into one complex. Fusion proteins are created by ligation of two or more protein domains in a single polypeptide with functional properties. Improvement of therapeutic function is one of the main goals for developing these products. Elimination from the body is one of the most important points that should be considered in the design and production of fusion proteins. This review describes some of the most important excretion/elimination pathways of fusion peptide and protein drugs as well as serious elimination challenges in the development and manufacturing of fusion proteins. Keywords: Fusion proteins; Therapy; Pharmacokinetics; Serum half-life; Peptide. -
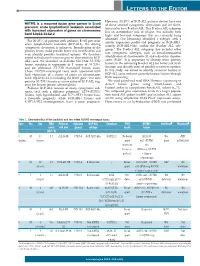
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds.