MASTER NATIONAL ACADEMY PRESS Washington, D.C 1983
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Early Synchrotrons in Britain, and Early Work for Cern
EARLY SYNCHROTRONS IN BRITAIN, AND EARLY WORK FOR CERN J. D. Lawson Formerly Rutherford Appleton Laboratory, Chilton, Oxon, UK Abstract Early work on electron synchrotrons in the UK, including an account of the conversion of a small betatron in 1946 to become the world’s first synchrotron, is described first. This is followed by a description of the design and construction of the 1 GeV synchrotron at the University of Birmingham which was started in the same year. Finally an account is given of the work of the international team during 1952–3, which formed the basis for the design of the CERN PS before the move to Geneva. It was during this year that John Adams showed the outstanding ability that later brought the project to such a successful conclusion. 1 EARLY PLANS IN BRITAIN: THE WORLD’S FIRST SYNCHROTRON During the second world war Britain’s nuclear physicists were deployed in research directed towards winning the war. Many were engaged in developments associated with radar, (or ‘radiolocation’ as it was then called), both at universities and at government laboratories, such as the radar establishments TRE and ADRDE at Malvern. Others contributed to the atomic bomb programme, both in the UK, and in the USA. Towards the end of the war, when victory seemed assured, the nuclear physicists began looking towards the peacetime future. The construction of new particle accelerators to achieve ever higher energies was seen as one of the more important possibilities. Those working at Berkeley on the electromagnetic separator were familiar with the accelerators there, and following the independent invention (or discovery?) there of the principle of phase stability by Edwin McMillan in 1945, exciting possibilities were immediately apparent [4]. -
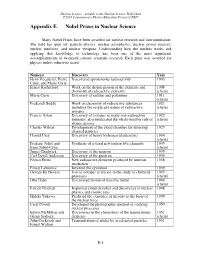
Appendix E Nobel Prizes in Nuclear Science
Nuclear Science—A Guide to the Nuclear Science Wall Chart ©2018 Contemporary Physics Education Project (CPEP) Appendix E Nobel Prizes in Nuclear Science Many Nobel Prizes have been awarded for nuclear research and instrumentation. The field has spun off: particle physics, nuclear astrophysics, nuclear power reactors, nuclear medicine, and nuclear weapons. Understanding how the nucleus works and applying that knowledge to technology has been one of the most significant accomplishments of twentieth century scientific research. Each prize was awarded for physics unless otherwise noted. Name(s) Discovery Year Henri Becquerel, Pierre Discovered spontaneous radioactivity 1903 Curie, and Marie Curie Ernest Rutherford Work on the disintegration of the elements and 1908 chemistry of radioactive elements (chem) Marie Curie Discovery of radium and polonium 1911 (chem) Frederick Soddy Work on chemistry of radioactive substances 1921 including the origin and nature of radioactive (chem) isotopes Francis Aston Discovery of isotopes in many non-radioactive 1922 elements, also enunciated the whole-number rule of (chem) atomic masses Charles Wilson Development of the cloud chamber for detecting 1927 charged particles Harold Urey Discovery of heavy hydrogen (deuterium) 1934 (chem) Frederic Joliot and Synthesis of several new radioactive elements 1935 Irene Joliot-Curie (chem) James Chadwick Discovery of the neutron 1935 Carl David Anderson Discovery of the positron 1936 Enrico Fermi New radioactive elements produced by neutron 1938 irradiation Ernest Lawrence -

Regional Oral History Office University of California the Bancroft Library Berkeley, California
Regional Oral History Office University of California The Bancroft Library Berkeley, California Program in Bioscience and Biotechnology Studies RONALD E. CAPE, M.B.A., Ph. D. BIOTECH PIONEER AND CO-FOUNDER OF CETUS Interviews Conducted by Sally Smith Hughes in 2003 Copyright © 2006 by The Regents of the University of California Since 1954 the Regional Oral History Office has been interviewing leading participants in or well-placed witnesses to major events in the development of northern California, the West, and the nation. Oral history is a method of collecting historical information through tape-recorded interviews between a narrator with firsthand knowledge of historically significant events and a well-informed interviewer, with the goal of preserving substantive additions to the historical record. The tape recording is transcribed, lightly edited for continuity and clarity, and reviewed by the interviewee. The corrected manuscript is indexed, bound with photographs and illustrative materials, and placed in The Bancroft Library at the University of California, Berkeley, and in other research collections for scholarly use. Because it is primary material, oral history is not intended to present the final, verified, or complete narrative of events. It is a spoken account, offered by the interviewee in response to questioning, and as such it is reflective, partisan, deeply involved, and irreplaceable. ************************************ All uses of this manuscript are covered by legal agreements between The Regents of the University of California and Ronald Cape, dated December 18, 2003. The manuscript is thereby made available for research purposes. All literary rights in the manuscript, including the right to publish, are reserved to The Bancroft Library of the University of California, Berkeley. -

Edwin M. Mcmillan, a Biographical Sketch
LBL 35965 HQDFAN668 EDWIN M. MCMILLAN, A BIOGRAPHICAL SKETCH EDWARD J. LOFGREN, Associate Director Emeritus LAWRENCE BERKELEY LABORATORY BERKELEY, CALIFORNIA 94720 Presented at the International Symposium: The 50th Anniversary of the Phase Stability Principle Moscow/Dubna, Russia 12-15 July 1994 ABSTRACT. Edwin M. McMillan was one of the great scientists of the middle years of this century. He made notable contributions to nuclear, and particle physics, the chemis try of transuranic elements, and accelerator physics. * This work supported by the Director, Office of Energy Research, Office of Fusion Energy, U.S. Department of Energy under contract No. DE-AC03-76SF00098. ^-TWBUTION OF THIS DOCUMENT (S UNLIMITED Edwin McMillan was born on Septem tion Laboratory. He joined the Laboratory in ber 18,1907 at Redondo Beach, California. Soon 1934 and began a lifelong close association with after, the family moved to Pasadena, California Lawrence. where the father practised medicine for many He quickly established himself as a me years. The move to Pasadena was a very fortu ticulous and versatile experimenter in nuclear nate one because Caltech was nearby and even physics with an excellent grasp of theory. Dur in his early school years Ed could take advan ing this period he discovered *-0 with Stanley tage of the excellent public programs and lec Livingston and 10Be with Samuel Ruben. Per tures put on there by them. This contact with the haps his best experiment at that time was the world of science as Ed grew from boyhood to a demonstration of electron pair production by the young man was very important, for it nurtured absorption of gamma rays from fluorine bom the wide and lasting curiosity and interest that barded by protons from the cyclotron. -
Nobel Laureate Glenn T. Seaborg Honored with Endowed Chair; Chemistry Professor Alexander Pines Named Chairholder
04.20.98 - Nobel Laureate Glenn T. Seaborg honored with end... http://www.berkeley.edu/news/media/releases/98legacy/04_20_... NEWS RELEASE, 04/20/98 Nobel Laureate Glenn T. Seaborg honored with endowed chair; chemistry Professor Alexander Pines named chairholder By Jane Scheiber BERKELEY -- Nobel Laureate Glenn T. Seaborg, who last year became the first living scientist to have an element named for him, has been honored again, this time with the establishment of the Glenn T. Seaborg Chair in Physical Chemistry at the University of California, Berkeley. The Chair was created through a generous endowment in tribute to Seaborg and the College of Chemistry. Endowed chairs, which provide a source of income to advance the scholarly activities of the appointed professor, also are one of the highest forms of campus recognition that a faculty member can receive. Alexander Pines, who has significantly advanced the field of nuclear magnetic resonance (NMR) spectroscopy, has been named to hold the Seaborg Chair. "It is a privilege indeed to be thus associated with my legendary colleague and good friend Glenn Seaborg," Pines says. "It is especially fitting that Glenn Seaborg, who has received the highest awards of the international and national scientific communities, be thus recognized at the institution where he earned his doctorate and where he has spent most of his career," said College of Chemistry Dean Alexis T. Bell. "We are enormously grateful to our anonymous benefactors." Bell added, "We anticipate that we will be able to recognize other distinguished faculty members as additional chairs are established in the future." Seaborg earned his Ph.D. -

Edwin Mcmillan Spent a Large Part of His Professional Life in Close Association with Ernest O
NATIONAL ACADEMY OF SCIENCES E D W I N M A T T I S O N M CMILLAN 1907—1991 A Biographical Memoir by J . DAVID JACKSON AN D W . K .H. PANOFSKY Any opinions expressed in this memoir are those of the author(s) and do not necessarily reflect the views of the National Academy of Sciences. Biographical Memoir COPYRIGHT 1996 NATIONAL ACADEMIES PRESS WASHINGTON D.C. Courtesy of the Lawrence Berkeley Laboratory EDWIN MATTISON MCMILLAN September 18, 1907–September 8, 1991 BY J. DAVID JACKSON AND W. K. H. PANOFSKY ITH THE DEATH OF Edwin Mattison McMillan on Sep- Wtember 8, 1991, the world lost one of its great natural scientists. We advisedly use the term “natural scientist” since McMillan’s interests transcended greatly that of his profes- sion of physicist. They encompassed everything natural from rocks through elementary particles to pure mathematics and included an insatiable appetite for understanding ev- erything from fundamental principles. Edwin McMillan spent a large part of his professional life in close association with Ernest O. Lawrence1 and succeeded Lawrence as director of what is now the Lawrence Berkeley Laboratory in 1958. Yet the two men could hardly be more different. Lawrence was a man of great intuition, outgoing, and a highly capable organizer of the work of many people. Edwin McMillan was thoroughly analytical in whatever he did and usually worked alone or with few associates. He disliked specialization and the division of physics divided into theory and experiment. He remarked at an interna- tional high-energy physics meeting, “Any experimentalist, unless proven a damn fool, should be given one half year to interpret his own experiment.” McMillan’s first and last publications illustrate the un- usual breadth of his interests. -

Chapter 4 Artificial Radioactive Isotopes
Irene and Frederic Joliot-Curie Chapter 4 Artificial Radioactive Isotopes The Discovery It was mentioned earlier that Irene Joliot-Curie and her husband Frederic Joliot-Curie was awarded the Nobel prize in Chemistry in 1935. The prize was given ”in recognition of their synthesis of new ra- dioactive elements” . Let us see more into the details of this experiment. Irène and Frederic Joliot-Curie had a large supply of polonium, after Irene`s parents. The polonium emitted alpha particles which they used to bombard different elements. In 1933 they used alpha particles and bombarded an aluminum plate. When they removed the α−particle source, it appeared that the aluminum plate emitted radiation with a half-life of approximately 3 minutes. The explanation was that the bombardment had resulted in a nuclear reaction. The α-particle penetrated the aluminum nucleus and changed it into phosphorus by emitting a neutron. The new phosphorus isotope was radioactive and was responsible for the observed radiation. Its designation is P-30. This nuclear reaction may be written as follows: 27 + α ⇒ 30 27 4 30 1 Al P + n or like this 13Al+ 2 He ⇒+ 15 P 0 n Decay of P-30 is: 30 30 15P⇒+ 14 Si positron Energy 3.24 MeV The neutron emitted can be observed as long as the bombardment takes place, but disappears im- mediately when the α-source is removed. However, the phosphorus isotope is radioactive. The decay mode is positron emission as shown above. The half-life is 2.5 minutes. 43 Irene and Fredric Joliot-Curie used their alpha bombardment technique on some other elements and found that it was possible to transform an element into another, with a higher number of protons in its nucleus. -

History of the Seaborg Institute
HISTORY OF THE SEABORG INSTITUTE ARQ.2_09.cover.indd 1 6/22/09 9:24 AM ContentsContents History of the Seaborg Institute The Seaborg Institute for Transactinium Science Established to educate the next generation of nuclear scientists 1 ITS Advisory Council 5 Seaborg the scientist 7 Naming seaborgium 11 Branching out 13 IGCAR honors Seaborg’s contributions to actinide science 16 Seaborg Institute for Transactinium Science/Los Alamos National Laboratory Actinide Research Quarterly The Seaborg Institute for Transactinium Science Established to educate the next generation of nuclear scientists In the late 1970s and early 1980s, research opportunities in heavy element This article was contributed by science and engineering were seldom found in university settings, and the supply Darleane C. Hoffman, professor emerita, of professionals in those fields was diminishing to the detriment of national Graduate School, Department of goals. In response to these concerns, national studies were conducted and panels Chemistry, UC Berkeley, faculty senior and committees were formed to assess the status of training and education in the scientist, Lawrence Berkeley National nuclear sciences. The studies in particular addressed the future need for scientists Laboratory, and charter director, Seaborg trained in the areas of nuclear waste management and disposal, environmental Institute, Lawrence Livermore National remediation, nuclear fuel processing, and nuclear safety analysis. Laboratory; and Christopher Gatrousis, Unfortunately, with the exception of the American Chemical Society’s former associate director of Chemistry Division of Nuclear Chemistry and Technology summer schools for undergradu- and Materials Science, Lawrence ates in nuclear and radiochemistry established in 1984, the studies did not lead Livermore National Laboratory. -

Segr㨠(Emilio)
http://oac.cdlib.org/findaid/ark:/13030/c8639vx8 No online items Finding Aid to the Emilio Segrè papers BANC MSS 78/72 cp Marjorie Bryer The Bancroft Library 2017 The Bancroft Library University of California Berkeley, CA 94720-6000 [email protected] URL: http://www.lib.berkeley.edu/libraries/bancroft-library Finding Aid to the Emilio Segrè BANC MSS 78/72 cp 1 papers BANC MSS 78/72 cp Language of Material: English Contributing Institution: The Bancroft Library Title: Segrè (Emilio) papers Creator: Segre, Emilio Identifier/Call Number: BANC MSS 78/72 cp Physical Description: 60 Linear Feet(40 cartons, 2 card file boxes, 1 oversize box, 3 oversize folders, 1 tube) Date (inclusive): 1870-1998, bulk 1939-1989 Date (bulk): 1939-1989 Abstract: This collection documents the personal and professional life of Nobel Prize-winning physicist and University of California, Berkeley professor Emilio Segrè and offers insights into the history of physics and physicists in the 20th Century. Segrè's papers include personal and professional correspondence; family papers and personalia; materials related to Segrè’s mentor and colleague, Enrico Fermi; articles, drafts, manuscripts, talks, and publications; journals and notebooks; book projects; records from the Lawrence Berkeley Radiation Lab and Los Alamos National laboratory; materials related to Segrè’s Nobel Prize; administrative records from the University of California Berkeley; course materials; and works by other physicists. Language of Material: Collection materials are in English, Italian, German and Russian. Many of the Bancroft Library collections are stored offsite and advance notice may be required for use. For current information on the location of these materials, please consult the library's online catalog. -
List of Nobel Laureates 1
List of Nobel laureates 1 List of Nobel laureates The Nobel Prizes (Swedish: Nobelpriset, Norwegian: Nobelprisen) are awarded annually by the Royal Swedish Academy of Sciences, the Swedish Academy, the Karolinska Institute, and the Norwegian Nobel Committee to individuals and organizations who make outstanding contributions in the fields of chemistry, physics, literature, peace, and physiology or medicine.[1] They were established by the 1895 will of Alfred Nobel, which dictates that the awards should be administered by the Nobel Foundation. Another prize, the Nobel Memorial Prize in Economic Sciences, was established in 1968 by the Sveriges Riksbank, the central bank of Sweden, for contributors to the field of economics.[2] Each prize is awarded by a separate committee; the Royal Swedish Academy of Sciences awards the Prizes in Physics, Chemistry, and Economics, the Karolinska Institute awards the Prize in Physiology or Medicine, and the Norwegian Nobel Committee awards the Prize in Peace.[3] Each recipient receives a medal, a diploma and a monetary award that has varied throughout the years.[2] In 1901, the recipients of the first Nobel Prizes were given 150,782 SEK, which is equal to 7,731,004 SEK in December 2007. In 2008, the winners were awarded a prize amount of 10,000,000 SEK.[4] The awards are presented in Stockholm in an annual ceremony on December 10, the anniversary of Nobel's death.[5] As of 2011, 826 individuals and 20 organizations have been awarded a Nobel Prize, including 69 winners of the Nobel Memorial Prize in Economic Sciences.[6] Four Nobel laureates were not permitted by their governments to accept the Nobel Prize. -

Explorerof the Mysteries of the Atom @1 VI Tions from the Faculty and Graduate Students on the Lat €˜U Est Research
Nuclear Medicine Pioneer: Glenn T. Seaborg w z Explorerof the Mysteries of the Atom @1 VI tions from the faculty and graduate students on the lat ‘U est research. Seaborg's col z 4 leagues and future friends included cyclotron inventor and future Nobel laureate Ernest 0. Lawrence, Robert Oppenheimer and future col laborators John (Jack) Livin I good and Edwin McMillan. Radioisotope Discoveries After completing his grad nate studies, Seaborg became 111 @; ..@ the personal research assistant @ GilbertN. Lewis, UCB'Sdean ofthe college of chemistry. @ (Left) Glenn 1. Seaberg A codiscoverer ofone ofthe most frequently While Seaborg assisted Lewis with his research on andEdwinMcMlIlanon used radionuclides in nuclear medicine, generalized acids andbases, duringhis sparetime, thedaytheywere notified they had won 99mTc,Nobel laureate Glenn T. Seaborg, generally at night, he continued his own investi theNobelPrize PhD, has contributed enormously to the specialty gations. His research was spurred when Livingood, (October 1951). (Right) ofnuclear medicine and created a firm foundation his codiscoverer of 1311,handed him a “hot―target Glenn T.Seaborg and for the development ofnuclear chemistry.The dis and asked him to chemically identify the radioiso Emilie Segrèpresenting topes itcontained.This initialforaywas the launch a sampleofplutonium coverer often elements, including one that bears totheMuseumof his name, Seaborg continues to draw on a lifetime ing point for Seaborg's life work and was the start History and Technology, ofexperience to advocate the safe uses of nuclear ofa 5-year collaboration with Livingood. In addi Smithsonian Institution, power and the need to invest in education and sup tion to 131!,the Seaborg-Livingood team also dis Washington, DC, March port of basic research and to pass on to future gen covered 59Feand 6OCoThe development of 131! 28, 1966. -

Character List
Character List - Bomb Use this chart to help you keep track of the hundreds of names of physicists, freedom fighters, government officials, and others involved in the making of the atomic bomb. Scientists Political/Military Leaders Spies Robert Oppenheimer - Winston Churchill -- Prime Klaus Fuchs - physicist in designed atomic bomb. He was Minister of England Manhattan Project who gave accused of spying. secrets to Russia Franklin D. Roosevelt -- Albert Einstein - convinced President of the United States Harry Gold - spy and Courier U.S. government that they for Russia KGB. Narrator of the needed to research fission. Harry Truman -- President of story the United States Enrico Fermi - created first Ruth Werner - Russian spy chain reaction Joseph Stalin -- dictator of the Tell Hall -- physicist in Soviet Union Igor Korchatov -- Russian Manhattan Project who gave physicist in charge of designing Adolf Hitler -- dictator of secrets to Russia bomb Germany Haakon Chevalier - friend who Werner Reisenberg -- Leslie Groves -- Military approached Oppenheimer about German physicist in charge of leader of the Manhattan Project spying for Russia. He was designing bomb watched by the FBI, but he was not charged. Otto Hahn -- German physicist who discovered fission Other scientists involved in the Manhattan Project: Aage Niels Bohr George Kistiakowsky Joseph W. Kennedy Richard Feynman Arthur C. Wahl Frank Oppenheimer Joseph Rotblat Robert Bacher Arthur H. Compton Hans Bethe Karl T. Compton Robert Serber Charles Critchfield Harold Agnew Kenneth Bainbridge Robert Wilson Charles Thomas Harold Urey Leo James Rainwater Rudolf Pelerls Crawford Greenewalt Harold DeWolf Smyth Leo Szilard Samuel K. Allison Cyril S. Smith Herbert L. Anderson Luis Alvarez Samuel Goudsmit Edward Norris Isidor I.